

Functional single nucleotide polymorphism-based association studies
- PMID: 16848977
- PMCID: PMC3525158
- DOI: 10.1186/1479-7364-2-6-391
Functional single nucleotide polymorphism-based association studies
Abstract
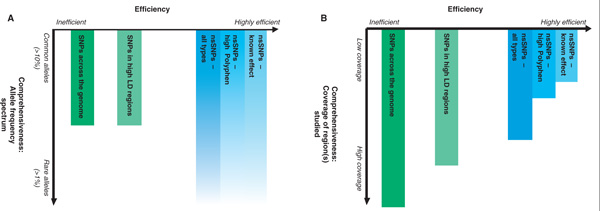
Association studies hold great promise for the elucidation of the genetic basis of diseases. Studies based on functional single nucleotide polymorphisms (SNPs) or on linkage disequilibrium (LD) represent two main types of designs. LD-based association studies can be comprehensive for common causative variants, but they perform poorly for rare alleles. Conversely, functional SNP-based studies are efficient because they focus on the SNPs with the highest a priori chance of being associated. Our poor ability to predict the functional effect of SNPs, however, hampers attempts to make these studies comprehensive. Recent progress in comparative genomics, and evidence that functional elements tend to lie in conserved regions, promises to change the landscape, permitting functional SNP association studies to be carried out that comprehensively assess common and rare alleles. SNP genotyping technologies are already sufficient for such studies, but studies will require continued genomic sequencing of multiple species, research on the functional role of conserved sequences and additional SNP discovery and validation efforts (including targeted SNP discovery to identify the rare alleles in functional regions). With these resources, we expect that comprehensive functional SNP association studies will soon be possible.
Figures
Similar articles
-
Single-nucleotide polymorphism genotyping for disease association studies.Methods Mol Med. 2005;108:159-72. doi: 10.1385/1-59259-850-1:159. Methods Mol Med. 2005. PMID: 16028683
-
The usefulness of different density SNP maps for disease association studies of common variants.Hum Mol Genet. 2003 Dec 1;12(23):3145-9. doi: 10.1093/hmg/ddg337. Epub 2003 Oct 7. Hum Mol Genet. 2003. PMID: 14532327
-
SNPs in Multi-species Conserved Sequences (MCS) as useful markers in association studies: a practical approach.BMC Genomics. 2007 Aug 6;8:266. doi: 10.1186/1471-2164-8-266. BMC Genomics. 2007. PMID: 17683615 Free PMC article.
-
SNP web resources and their potential applications in personalized medicine.Curr Drug Metab. 2012 Sep 1;13(7):978-90. doi: 10.2174/138920012802138552. Curr Drug Metab. 2012. PMID: 22591348 Review.
-
Applications of single nucleotide polymorphisms in crop genetics.Curr Opin Plant Biol. 2002 Apr;5(2):94-100. doi: 10.1016/s1369-5266(02)00240-6. Curr Opin Plant Biol. 2002. PMID: 11856602 Review.
Cited by
-
Genetic architecture of hippocampus subfields volumes in Alzheimer's disease.CNS Neurosci Ther. 2024 Mar;30(3):e14110. doi: 10.1111/cns.14110. Epub 2023 Feb 8. CNS Neurosci Ther. 2024. PMID: 36756718 Free PMC article.
-
Identification of SNPs associated with muscle yield and quality traits using allelic-imbalance analyses of pooled RNA-Seq samples in rainbow trout.BMC Genomics. 2017 Aug 7;18(1):582. doi: 10.1186/s12864-017-3992-z. BMC Genomics. 2017. PMID: 28784089 Free PMC article.
-
The association of GATM polymorphism with statin-induced myopathy: a systematic review and meta-analysis.Eur J Clin Pharmacol. 2021 Mar;77(3):349-357. doi: 10.1007/s00228-020-03019-3. Epub 2020 Oct 13. Eur J Clin Pharmacol. 2021. PMID: 33051696 Free PMC article.
-
Low pyrrolizidine alkaloid levels in perennial ryegrass is associated with the absence of a homospermidine synthase gene.BMC Plant Biol. 2018 Apr 6;18(1):56. doi: 10.1186/s12870-018-1269-6. BMC Plant Biol. 2018. PMID: 29625552 Free PMC article.
-
Effect of HPSE and HPSE2 SNPs on the Risk of Developing Primary Paraskeletal Multiple Myeloma.Cells. 2023 Mar 16;12(6):913. doi: 10.3390/cells12060913. Cells. 2023. PMID: 36980254 Free PMC article.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
