

Protein-protein interactions more conserved within species than across species
- PMID: 16854211
- PMCID: PMC1513270
- DOI: 10.1371/journal.pcbi.0020079
Protein-protein interactions more conserved within species than across species
Abstract
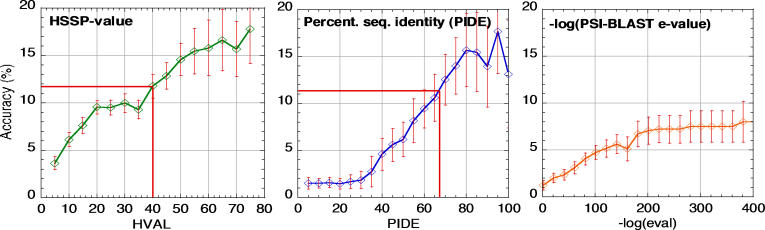
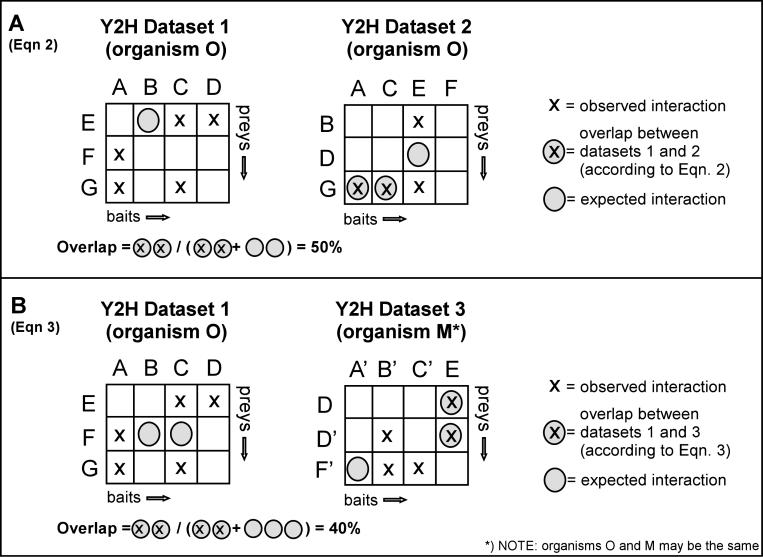
Experimental high-throughput studies of protein-protein interactions are beginning to provide enough data for comprehensive computational studies. Today, about ten large data sets, each with thousands of interacting pairs, coarsely sample the interactions in fly, human, worm, and yeast. Another about 55,000 pairs of interacting proteins have been identified by more careful, detailed biochemical experiments. Most interactions are experimentally observed in prokaryotes and simple eukaryotes; very few interactions are observed in higher eukaryotes such as mammals. It is commonly assumed that pathways in mammals can be inferred through homology to model organisms, e.g. the experimental observation that two yeast proteins interact is transferred to infer that the two corresponding proteins in human also interact. Two pairs for which the interaction is conserved are often described as interologs. The goal of this investigation was a large-scale comprehensive analysis of such inferences, i.e. of the evolutionary conservation of interologs. Here, we introduced a novel score for measuring the overlap between protein-protein interaction data sets. This measure appeared to reflect the overall quality of the data and was the basis for our two surprising results from our large-scale analysis. Firstly, homology-based inferences of physical protein-protein interactions appeared far less successful than expected. In fact, such inferences were accurate only for extremely high levels of sequence similarity. Secondly, and most surprisingly, the identification of interacting partners through sequence similarity was significantly more reliable for protein pairs within the same organism than for pairs between species. Our analysis underlined that the discrepancies between different datasets are large, even when using the same type of experiment on the same organism. This reality considerably constrains the power of homology-based transfer of interactions. In particular, the experimental probing of interactions in distant model organisms has to be undertaken with some caution. More comprehensive images of protein-protein networks will require the combination of many high-throughput methods, including in silico inferences and predictions. http://www.rostlab.org/results/2006/ppi_homology/
Conflict of interest statement
Figures





Similar articles
-
Annotation transfer between genomes: protein-protein interologs and protein-DNA regulogs.Genome Res. 2004 Jun;14(6):1107-18. doi: 10.1101/gr.1774904. Genome Res. 2004. PMID: 15173116 Free PMC article.
-
Evolutionary conservation of domain-domain interactions.Genome Biol. 2006;7(12):R125. doi: 10.1186/gb-2006-7-12-r125. Genome Biol. 2006. PMID: 17184549 Free PMC article.
-
Unequal evolutionary conservation of human protein interactions in interologous networks.Genome Biol. 2007;8(5):R95. doi: 10.1186/gb-2007-8-5-r95. Genome Biol. 2007. PMID: 17535438 Free PMC article.
-
Inferring protein-protein interactions from multiple protein domain combinations.Methods Mol Biol. 2009;541:43-59. doi: 10.1007/978-1-59745-243-4_3. Methods Mol Biol. 2009. PMID: 19381530 Review.
-
Origin of multicellular eukaryotes - insights from proteome comparisons.Curr Opin Genet Dev. 1999 Dec;9(6):688-94. doi: 10.1016/s0959-437x(99)00028-3. Curr Opin Genet Dev. 1999. PMID: 10607613 Review.
Cited by
-
The ortholog conjecture is untestable by the current gene ontology but is supported by RNA sequencing data.PLoS Comput Biol. 2012;8(11):e1002784. doi: 10.1371/journal.pcbi.1002784. Epub 2012 Nov 29. PLoS Comput Biol. 2012. PMID: 23209392 Free PMC article.
-
The ortholog conjecture revisited: the value of orthologs and paralogs in function prediction.Bioinformatics. 2020 Jul 1;36(Suppl_1):i219-i226. doi: 10.1093/bioinformatics/btaa468. Bioinformatics. 2020. PMID: 32657391 Free PMC article.
-
What evidence is there for the homology of protein-protein interactions?PLoS Comput Biol. 2012;8(9):e1002645. doi: 10.1371/journal.pcbi.1002645. Epub 2012 Sep 20. PLoS Comput Biol. 2012. PMID: 23028270 Free PMC article.
-
Host pathogen protein interactions predicted by comparative modeling.Protein Sci. 2007 Dec;16(12):2585-96. doi: 10.1110/ps.073228407. Epub 2007 Oct 26. Protein Sci. 2007. PMID: 17965183 Free PMC article.
-
Prediction of GCRV virus-host protein interactome based on structural motif-domain interactions.BMC Bioinformatics. 2017 Mar 2;18(1):145. doi: 10.1186/s12859-017-1500-8. BMC Bioinformatics. 2017. PMID: 28253857 Free PMC article.
References
-
- Fields S, Song O. A novel genetic system to detect protein–protein interactions. Nature. 1989;340:245–246. - PubMed
-
- Causier B. Studying the interactome with the yeast two-hybrid system and mass spectrometry. Mass Spectrom Rev. 2004;23:350–367. - PubMed
-
- Legrain P, Wojcik J, Gauthier JM. Protein–protein interaction maps: A lead towards cellular functions. Trends Genet. 2001;17:346–352. - PubMed
-
- Willats WG. Phage display: Practicalities and prospects. Plant Mol Biol. 2002;50:837–854. - PubMed
-
- Puig O, Caspary F, Rigaut G, Rutz B, Bouveret E, et al. The tandem affinity purification (TAP) method: A general procedure of protein complex purification. Methods. 2001;24:218–229. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
