

Extracting gene networks for low-dose radiation using graph theoretical algorithms
- PMID: 16854212
- PMCID: PMC1513268
- DOI: 10.1371/journal.pcbi.0020089
Extracting gene networks for low-dose radiation using graph theoretical algorithms
Abstract
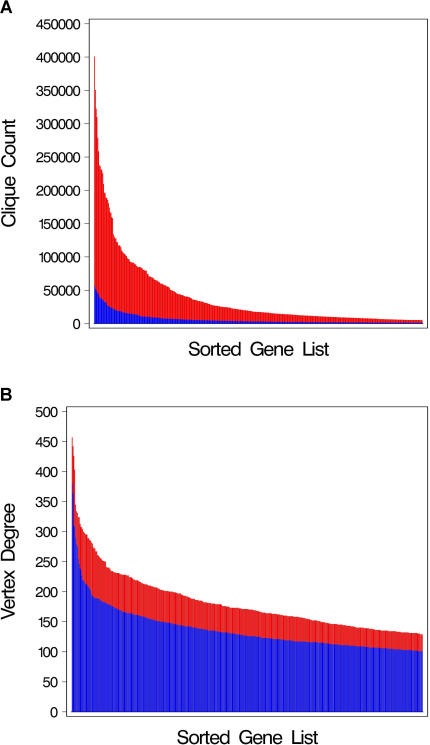
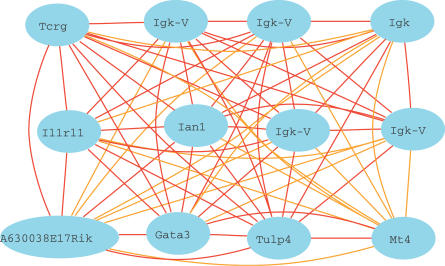
Genes with common functions often exhibit correlated expression levels, which can be used to identify sets of interacting genes from microarray data. Microarrays typically measure expression across genomic space, creating a massive matrix of co-expression that must be mined to extract only the most relevant gene interactions. We describe a graph theoretical approach to extracting co-expressed sets of genes, based on the computation of cliques. Unlike the results of traditional clustering algorithms, cliques are not disjoint and allow genes to be assigned to multiple sets of interacting partners, consistent with biological reality. A graph is created by thresholding the correlation matrix to include only the correlations most likely to signify functional relationships. Cliques computed from the graph correspond to sets of genes for which significant edges are present between all members of the set, representing potential members of common or interacting pathways. Clique membership can be used to infer function about poorly annotated genes, based on the known functions of better-annotated genes with which they share clique membership (i.e., "guilt-by-association"). We illustrate our method by applying it to microarray data collected from the spleens of mice exposed to low-dose ionizing radiation. Differential analysis is used to identify sets of genes whose interactions are impacted by radiation exposure. The correlation graph is also queried independently of clique to extract edges that are impacted by radiation. We present several examples of multiple gene interactions that are altered by radiation exposure and thus represent potential molecular pathways that mediate the radiation response.
Conflict of interest statement
Figures






Similar articles
-
k-Partite cliques of protein interactions: A novel subgraph topology for functional coherence analysis on PPI networks.J Theor Biol. 2014 Jan 7;340:146-54. doi: 10.1016/j.jtbi.2013.09.013. Epub 2013 Sep 19. J Theor Biol. 2014. PMID: 24056214
-
MICRAT: a novel algorithm for inferring gene regulatory networks using time series gene expression data.BMC Syst Biol. 2018 Dec 14;12(Suppl 7):115. doi: 10.1186/s12918-018-0635-1. BMC Syst Biol. 2018. PMID: 30547796 Free PMC article.
-
CoGA: An R Package to Identify Differentially Co-Expressed Gene Sets by Analyzing the Graph Spectra.PLoS One. 2015 Aug 27;10(8):e0135831. doi: 10.1371/journal.pone.0135831. eCollection 2015. PLoS One. 2015. PMID: 26313749 Free PMC article.
-
Transcriptional response of lymphoblastoid cells to ionizing radiation.Genome Res. 2003 Sep;13(9):2092-100. doi: 10.1101/gr.1240103. Epub 2003 Aug 12. Genome Res. 2003. PMID: 12915489 Free PMC article.
-
Identifying biological pathways via phase decomposition and profile extraction.Comput Syst Bioinformatics Conf. 2006:269-80. Comput Syst Bioinformatics Conf. 2006. PMID: 17369645
Cited by
-
Bridging the gap between systems biology and medicine.Genome Med. 2009 Sep 29;1(9):88. doi: 10.1186/gm88. Genome Med. 2009. PMID: 19754960 Free PMC article.
-
A Comparative Study of Gene Co-Expression Thresholding Algorithms.J Comput Biol. 2024 Jun;31(6):539-548. doi: 10.1089/cmb.2024.0509. Epub 2024 May 23. J Comput Biol. 2024. PMID: 38781420 Free PMC article.
-
Highly interconnected genes in disease-specific networks are enriched for disease-associated polymorphisms.Genome Biol. 2012 Jun 15;13(6):R46. doi: 10.1186/gb-2012-13-6-r46. Genome Biol. 2012. PMID: 22703998 Free PMC article.
-
Identification of innate immunity genes and pathways using a comparative genomics approach.Proc Natl Acad Sci U S A. 2008 May 13;105(19):7016-21. doi: 10.1073/pnas.0802405105. Epub 2008 May 7. Proc Natl Acad Sci U S A. 2008. PMID: 18463287 Free PMC article.
-
Using genomics to understand intestinal biology.J Physiol Biochem. 2007 Mar;63(1):83-96. doi: 10.1007/BF03174088. J Physiol Biochem. 2007. PMID: 17722646 Review.
References
-
- Wu LF, Hughes TR, Davierwala AP, Robinson MD, Stoughton R, et al. Large-scale prediction of Saccharomyces cerevisiae gene function using overlapping transcriptional clusters. Nat Genet. 2002;31:255–265. - PubMed
-
- Stuart JM, Segal E, Koller D, Kim SK. A gene-coexpression network for global discovery of conserved genetic modules. Science. 2003;302((5643)):249–255. - PubMed
-
- Hughes TR, Marton MJ, Jones AR, Roberts CJ, Stoughton R, et al. Functional discovery via a compendium of expression profiles. Cell. 2000;102:109–126. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
