

Microsomal prostaglandin E synthase-1 is a critical factor of stroke-reperfusion injury
- PMID: 16864802
- PMCID: PMC1518807
- DOI: 10.1073/pnas.0604400103
Microsomal prostaglandin E synthase-1 is a critical factor of stroke-reperfusion injury
Abstract
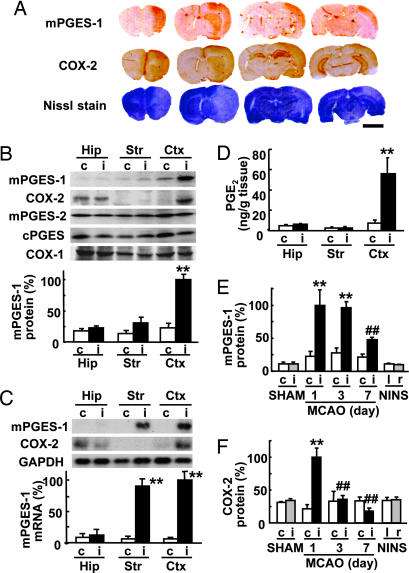
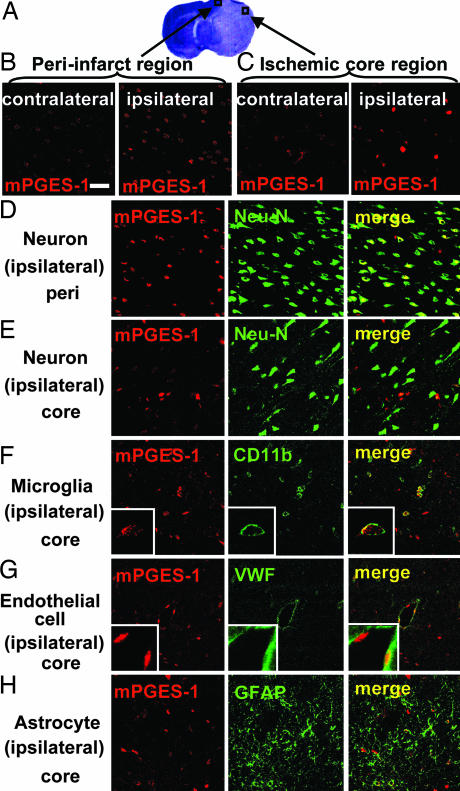
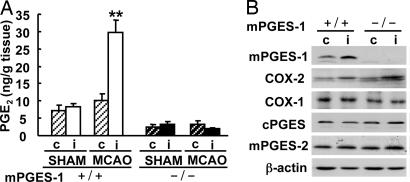
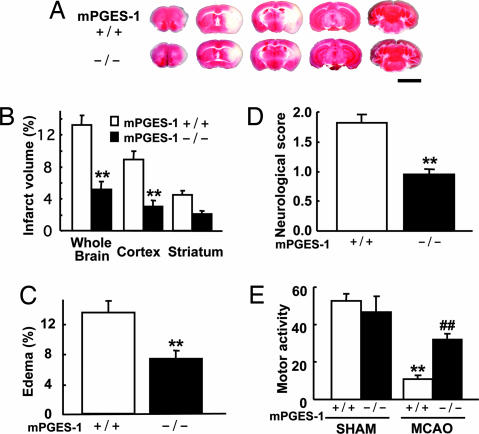
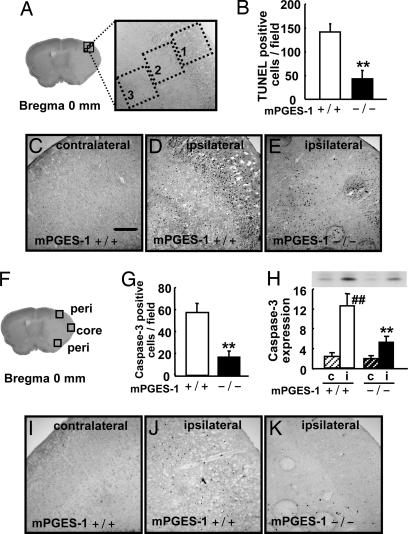
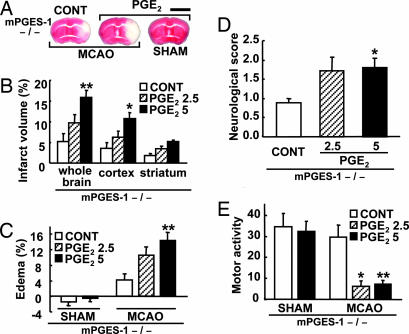
Although augmented prostaglandin E(2) (PGE(2)) synthesis and accumulation have been demonstrated in the lesion sites of rodent transient focal ischemia models, the role of PGE(2) in neuronal survival has been controversial, showing both protective and toxic effects. Here we demonstrate the induction of microsomal PGE synthase 1 (mPGES-1), an inducible terminal enzyme for PGE(2) synthesis, in neurons, microglia, and endothelial cells in the cerebral cortex after transient focal ischemia. In mPGES-1 knockout (KO) mice, in which the postischemic PGE(2) production in the cortex was completely absent, the infarction, edema, apoptotic cell death, and caspase-3 activation in the cortex after ischemia were all reduced compared with those in wild-type (WT) mice. Furthermore, the behavioral neurological dysfunctions observed after ischemia in WT mice were significantly ameliorated in KO mice. The ameliorated symptoms observed in KO mice after ischemia were reversed to almost the same severity as WT mice by intracerebroventricular injection of PGE(2) into KO mice. Our observations suggest that mPGES-1 may be a critical determinant of postischemic neurological dysfunctions and a valuable therapeutic target for treatment of human stroke.
Conflict of interest statement
Conflict of interest statement: No conflicts declared.
Figures
References
-
- Lyden P. D., Grotta J. C., Levine S. R., Marler J. R., Frankel M. R., Brott T. G. Neurology. 1997;49:14–20. - PubMed
-
- Lyden P. D. Prog. Cardiovasc. Dis. 1999;42:175–183. - PubMed
-
- Koistinaho J., Hokfelt T. NeuroReport. 1997;8:i–viii. - PubMed
-
- Dirnagl U., Iadecola C., Moskowitz M. A. Trends Neurosci. 1999;22:391–397. - PubMed
-
- Barone F. C., Feuerstein G. Z. J. Cereb. Blood Flow Metab. 1999;19:819–834. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
