

Molecular control of vertebrate iron homeostasis by iron regulatory proteins
- PMID: 16872694
- PMCID: PMC2291536
- DOI: 10.1016/j.bbamcr.2006.05.004
Molecular control of vertebrate iron homeostasis by iron regulatory proteins
Abstract
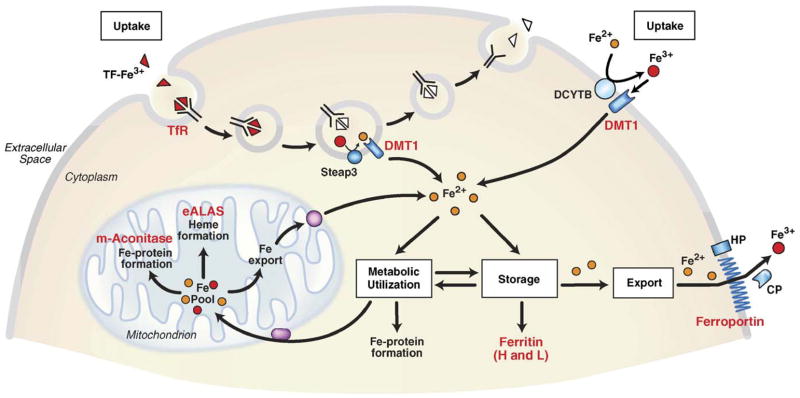
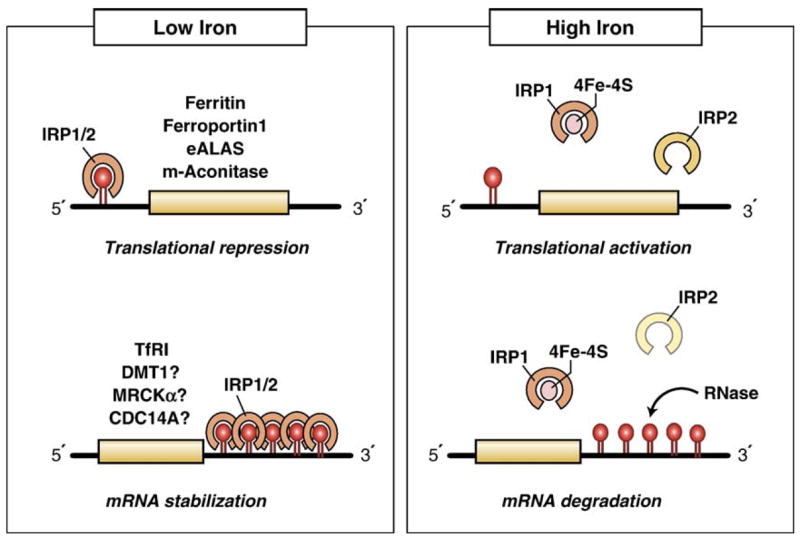
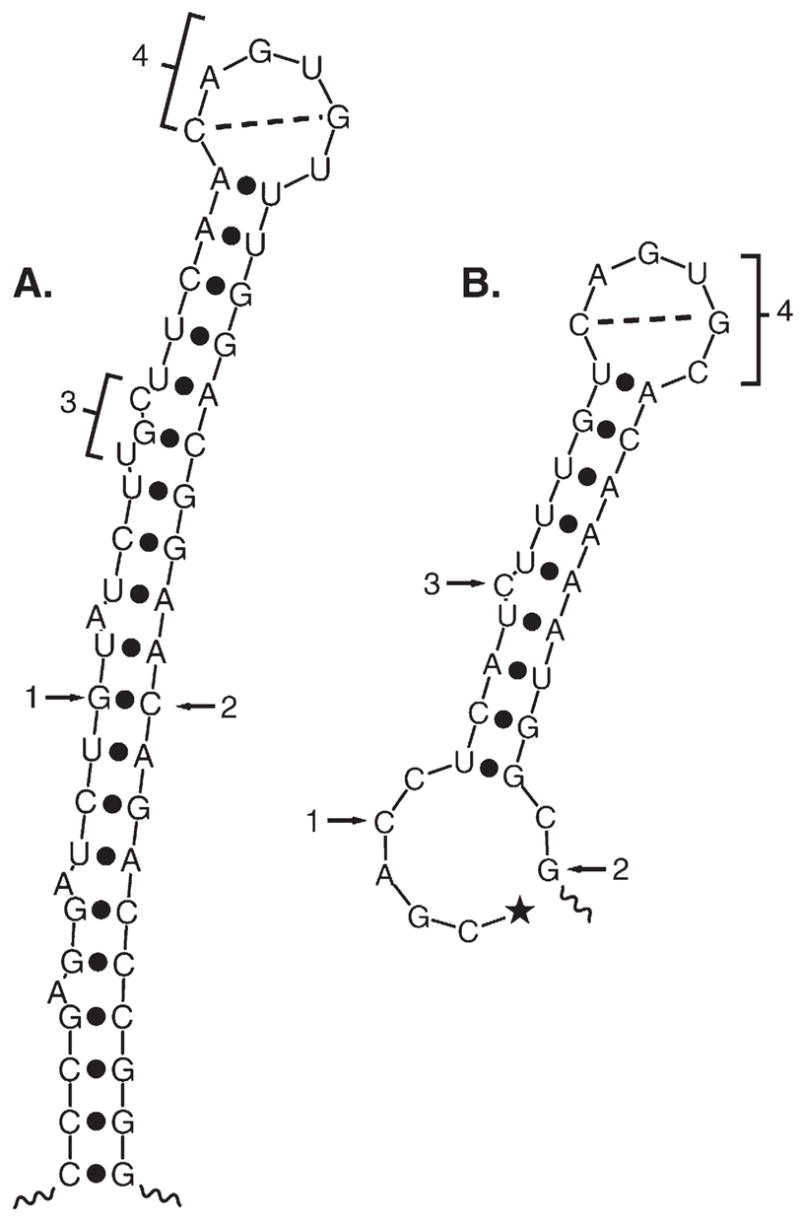
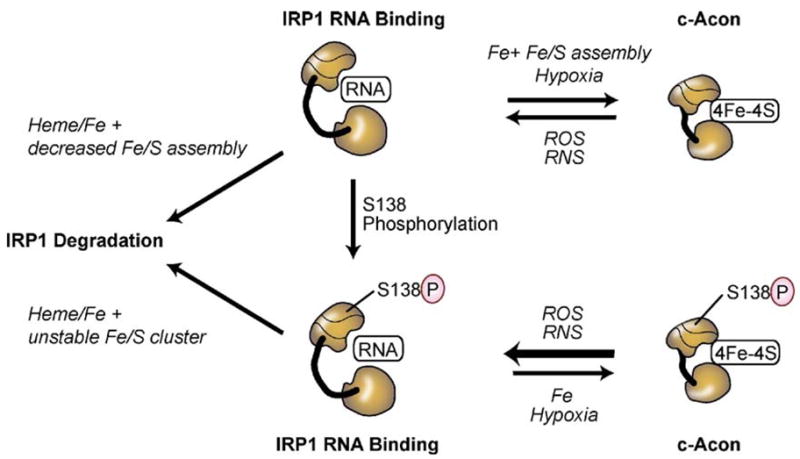
Both deficiencies and excesses of iron represent major public health problems throughout the world. Understanding the cellular and organismal processes controlling iron homeostasis is critical for identifying iron-related diseases and in advancing the clinical treatments for such disorders of iron metabolism. Iron regulatory proteins (IRPs) 1 and 2 are key regulators of vertebrate iron metabolism. These RNA binding proteins post-transcriptionally control the stability or translation of mRNAs encoding proteins involved in iron homeostasis thereby controlling the uptake, utilization, storage or export of iron. Recent evidence provides insight into how IRPs selectively control the translation or stability of target mRNAs, how IRP RNA binding activity is controlled by iron-dependent and iron-independent effectors, and the pathological consequences of dysregulation of the IRP system.
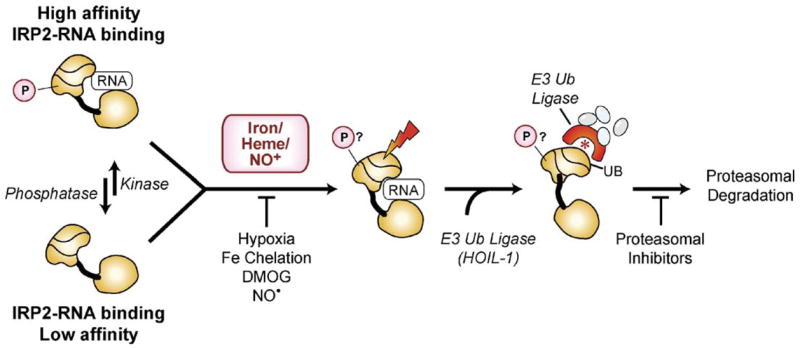
Figures
References
-
- Wood RJ, Ronnenberg AG. Iron. 10. Lippincott Williams and Wilkins; Philadelphia, PA: 2006.
-
- Lynch SR. The impact of iron fortification on nutritional anaemia. Best Pract Res Clin Haematol. 2005;18:333–346. - PubMed
-
- Hunt JM. Reversing productivity losses from iron deficiency: the economic case. J Nutr. 2002;132:794S–801S. - PubMed
-
- WHO. The World Health Report 2002-Reducing Risks, Promoting Health Life Styles. World Health Organization; 2002.
-
- Anonymous. Iron deficiency—United States, 1999–2000. MMWR. 2002;51:897–899. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
