

Proton-coupled electron transfer: the mechanistic underpinning for radical transport and catalysis in biology
- PMID: 16873123
- PMCID: PMC1647304
- DOI: 10.1098/rstb.2006.1874
Proton-coupled electron transfer: the mechanistic underpinning for radical transport and catalysis in biology
Abstract
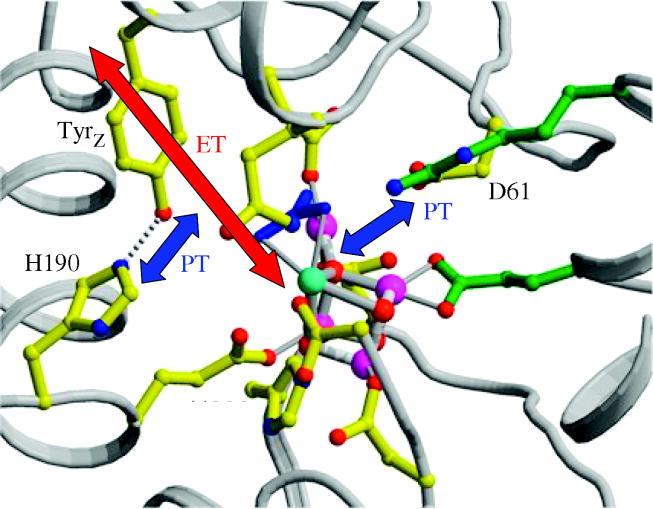
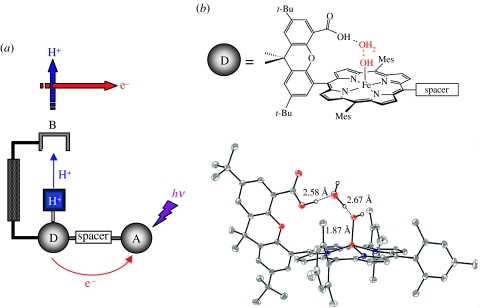
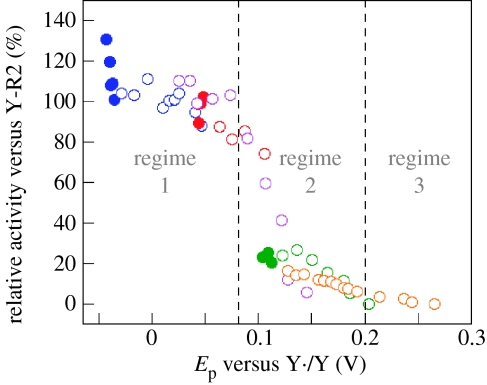
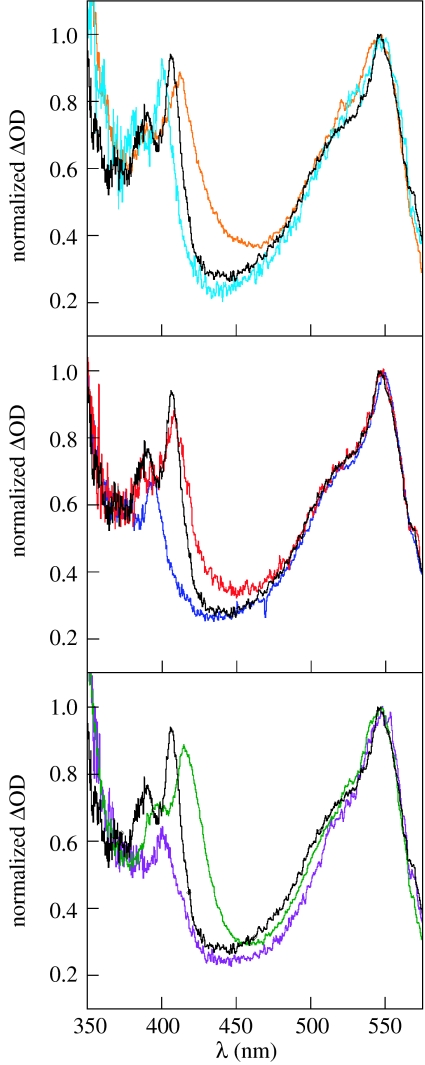
Charge transport and catalysis in enzymes often rely on amino acid radicals as intermediates. The generation and transport of these radicals are synonymous with proton-coupled electron transfer (PCET), which intrinsically is a quantum mechanical effect as both the electron and proton tunnel. The caveat to PCET is that proton transfer (PT) is fundamentally limited to short distances relative to electron transfer (ET). This predicament is resolved in biology by the evolution of enzymes to control PT and ET coordinates on highly different length scales. In doing so, the enzyme imparts exquisite thermodynamic and kinetic controls over radical transport and radical-based catalysis at cofactor active sites. This discussion will present model systems containing orthogonal ET and PT pathways, thereby allowing the proton and electron tunnelling events to be disentangled. Against this mechanistic backdrop, PCET catalysis of oxygen-oxygen bond activation by mono-oxygenases is captured at biomimetic porphyrin redox platforms. The discussion concludes with the case study of radical-based quantum catalysis in a natural biological enzyme, class I Escherichia coli ribonucleotide reductase. Studies are presented that show the enzyme utilizes both collinear and orthogonal PCET to transport charge from an assembled diiron-tyrosyl radical cofactor to the active site over 35A away via an amino acid radical-hopping pathway spanning two protein subunits.
Figures









References
-
- Aubert C, Vos M.H, Mathis P, Eker A.P.M, Brettel K. Intraprotein radical transfer during photoactivation of DNA photolyase. Nature. 2000;405:586–590. doi:10.1038/35014644 - DOI - PubMed
-
- Baldwin J, Krebs C, Ley B.A, Edmondson D.E, Huynh B.H, Bollinger J.M., Jr Mechanism of rapid electron transfer during oxygen activation in the R2 subunit of Escherichia coli ribonucleotide reductase. 1. Evidence for a transient tryptophan radical. J. Am. Chem. Soc. 2000;122:12 195–12 206. doi:10.1021/ja001278u - DOI
-
- Barber J, Ferreira K, Maghlaoui K, Iwata S. Structural model of the oxygen-evolving center of photosystem II with mechanistic implications. Phys. Chem. Chem. Phys. 2004;6:4737–4742. doi:10.1039/b407981g - DOI
-
- Barry B, Babcock G.T. Tyrosine radicals are involved in the photosynthetic oxygen-evolving system. Proc. Natl Acad. Sci. USA. 1987;84:7099–7103. doi:10.1073/pnas.84.20.7099 - DOI - PMC - PubMed
-
- Beratan D.N, Onuchic J.N, Hopfield J.J. Electron tunneling through covalent and noncovalent pathways in proteins. J. Chem. Phys. 1987;86:4488–4498. doi:10.1063/1.452723 - DOI
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
