

Recruitment of TFIIH to the HIV LTR is a rate-limiting step in the emergence of HIV from latency
- PMID: 16874302
- PMCID: PMC1538560
- DOI: 10.1038/sj.emboj.7601248
Recruitment of TFIIH to the HIV LTR is a rate-limiting step in the emergence of HIV from latency
Abstract
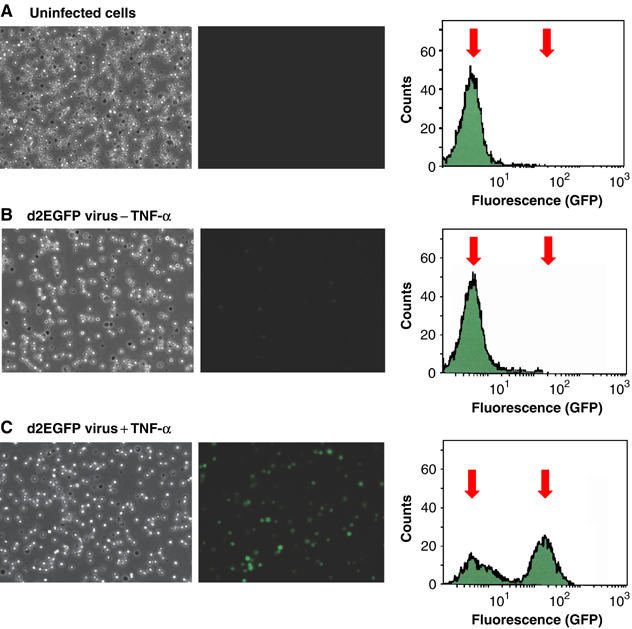
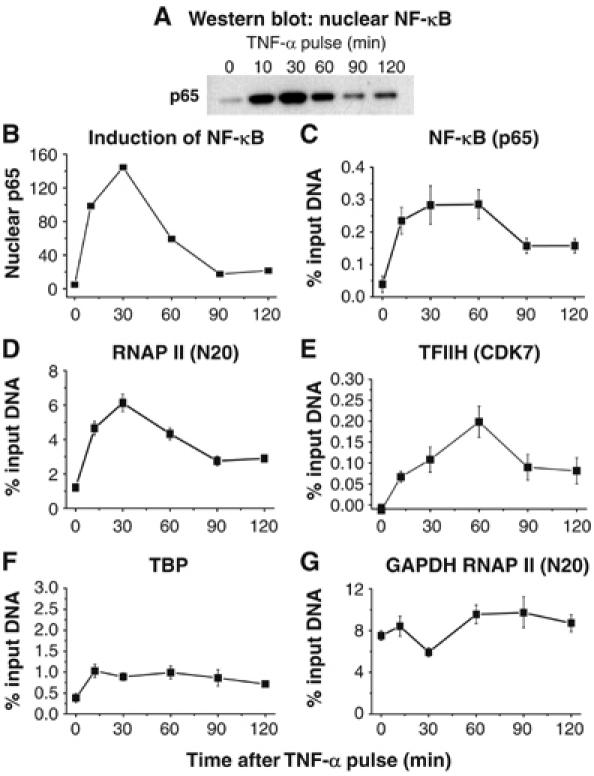
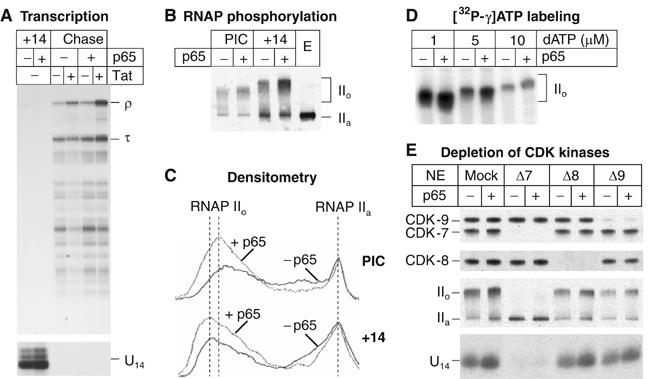
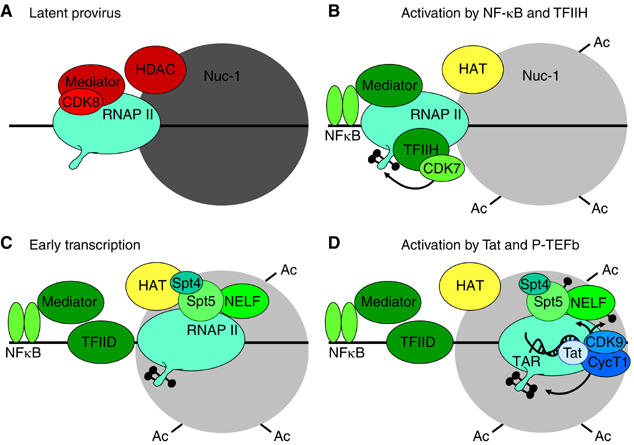
Latently infected cells rapidly initiate HIV transcription after exposure to signals that induce NF-kappaB. To investigate the role of TFIIH during HIV reactivation in vivo, we developed a population of Jurkat cells containing integrated, but transcriptionally silent, HIV proviruses. Surprisingly, the HIV promoter in unactivated Jurkat T cells is partially occupied and carries Mediator containing the CDK8 repressive module, TFIID and RNAP II that is hypophosphorylated and confined to the promoter region. Significantly, the promoter is devoid of TFIIH. Upon stimulation of the cells by TNF-alpha, NF-kappaB and TFIIH are rapidly recruited to the promoter together with additional Mediator and RNAP II, but CDK8 is lost. Detailed time courses show that the levels of TFIIH at the promoter fluctuate in parallel with NF-kappaB recruitment to the promoter. Similarly, recombinant p65 activates HIV transcription in vitro and stimulates phosphorylation of the RNAP II CTD by the CDK7 kinase module of TFIIH. We conclude that the recruitment and activation of TFIIH represents a rate-limiting step for the emergence of HIV from latency.
Figures



Similar articles
-
Negative elongation factor is required for the maintenance of proviral latency but does not induce promoter-proximal pausing of RNA polymerase II on the HIV long terminal repeat.Mol Cell Biol. 2014 Jun;34(11):1911-28. doi: 10.1128/MCB.01013-13. Epub 2014 Mar 17. Mol Cell Biol. 2014. PMID: 24636995 Free PMC article.
-
Naf1 Regulates HIV-1 Latency by Suppressing Viral Promoter-Driven Gene Expression in Primary CD4+ T Cells.J Virol. 2016 Dec 16;91(1):e01830-16. doi: 10.1128/JVI.01830-16. Print 2017 Jan 1. J Virol. 2016. PMID: 27795436 Free PMC article.
-
NF-kappaB p50 promotes HIV latency through HDAC recruitment and repression of transcriptional initiation.EMBO J. 2006 Jan 11;25(1):139-49. doi: 10.1038/sj.emboj.7600900. Epub 2005 Dec 1. EMBO J. 2006. PMID: 16319923 Free PMC article.
-
The essential and multifunctional TFIIH complex.Protein Sci. 2018 Jun;27(6):1018-1037. doi: 10.1002/pro.3424. Epub 2018 Apr 27. Protein Sci. 2018. PMID: 29664212 Free PMC article. Review.
-
Chromatin, gene silencing and HIV latency.Genome Biol. 2007;8(11):228. doi: 10.1186/gb-2007-8-11-228. Genome Biol. 2007. PMID: 18036274 Free PMC article. Review.
Cited by
-
H3K27 Demethylation at the Proviral Promoter Sensitizes Latent HIV to the Effects of Vorinostat in Ex Vivo Cultures of Resting CD4+ T Cells.J Virol. 2015 Aug;89(16):8392-405. doi: 10.1128/JVI.00572-15. Epub 2015 Jun 3. J Virol. 2015. PMID: 26041287 Free PMC article.
-
CBX4 contributes to HIV-1 latency by forming phase-separated nuclear bodies and SUMOylating EZH2.EMBO Rep. 2022 Jul 5;23(7):e53855. doi: 10.15252/embr.202153855. Epub 2022 Jun 1. EMBO Rep. 2022. PMID: 35642598 Free PMC article.
-
Selected drugs with reported secondary cell-differentiating capacity prime latent HIV-1 infection for reactivation.J Virol. 2012 Sep;86(17):9055-69. doi: 10.1128/JVI.00793-12. Epub 2012 Jun 13. J Virol. 2012. PMID: 22696646 Free PMC article.
-
Mechanisms of HIV Transcriptional Regulation and Their Contribution to Latency.Mol Biol Int. 2012;2012:614120. doi: 10.1155/2012/614120. Epub 2012 Jun 3. Mol Biol Int. 2012. PMID: 22701796 Free PMC article.
-
HIV Transcription Is Independent of Mediator Kinases.AIDS Res Hum Retroviruses. 2019 Aug;35(8):710-717. doi: 10.1089/AID.2019.0039. Epub 2019 May 29. AIDS Res Hum Retroviruses. 2019. PMID: 31044597 Free PMC article.
References
-
- Alcami J, de Lera TL, Folgueira L, Pedraza M-A, Jacqué J-M, Bachelerie F, Noriega AR, Hay RT, Harrich D, Gaynor RB, Virelizier J-L, Arenzana-Seisdedos F (1995) Absolute dependence on kB responsive elements for initiation and Tat-mediated amplification of HIV transcription in blood CD4T lymphocytes. EMBO J 14: 1552–1560 - PMC - PubMed
-
- Barboric M, Nissen RM, Kanazawa S, Jabrane-Ferrat N, Peterlin BM (2001) NF-kB binds P-TEFb to stimulate transcriptional elongation by RNA polymerase II. Mol Cell 8: 327–337 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
