

Cerebral ischemia-hypoxia induces intravascular coagulation and autophagy
- PMID: 16877357
- PMCID: PMC1780162
- DOI: 10.2353/ajpath.2006.051066
Cerebral ischemia-hypoxia induces intravascular coagulation and autophagy
Abstract
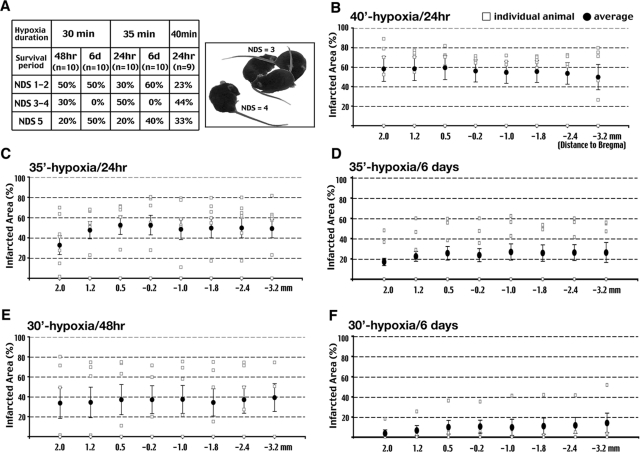
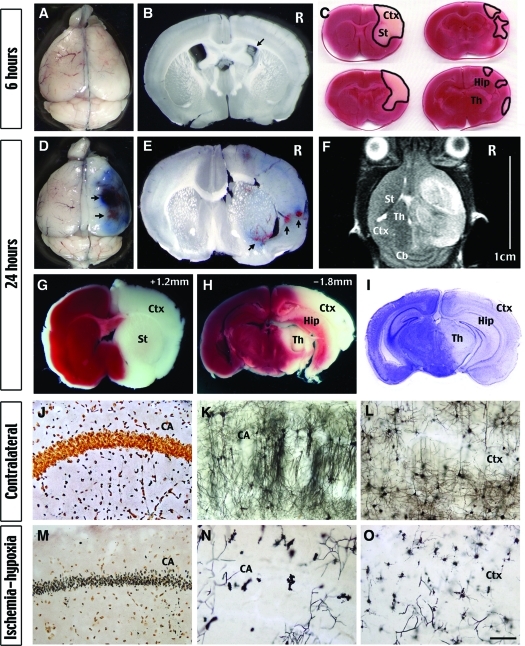
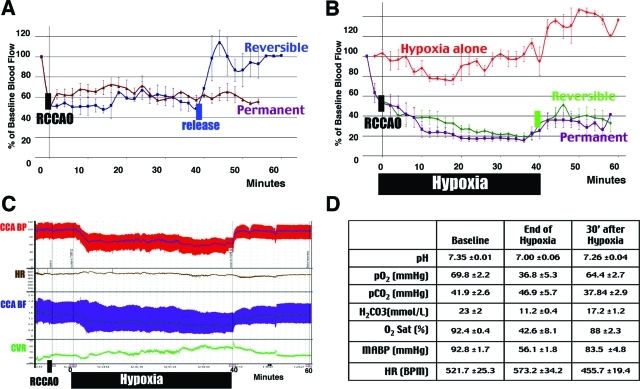
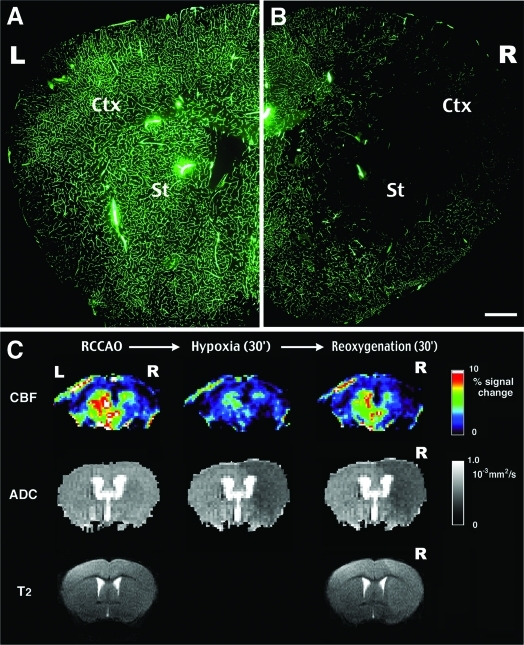
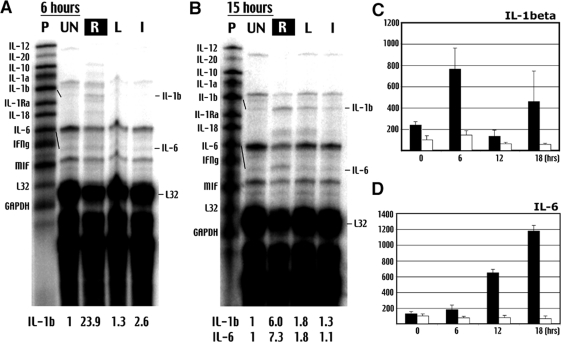
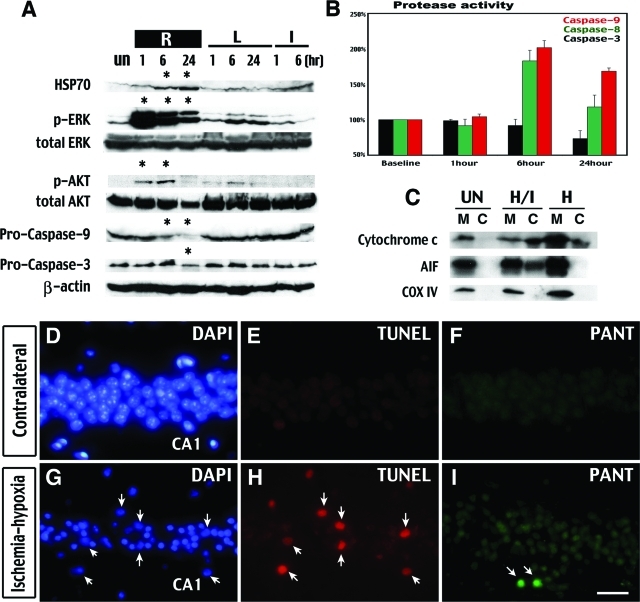
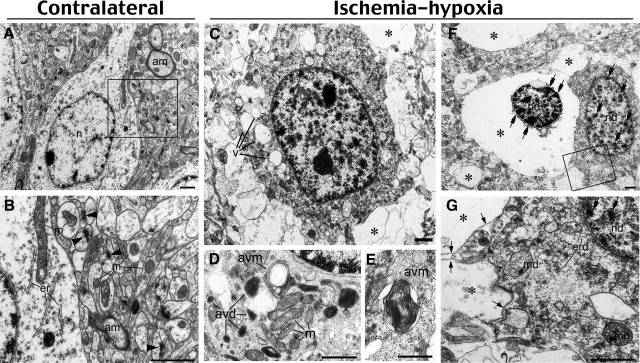
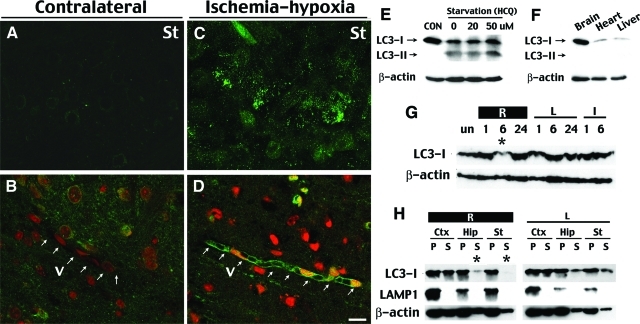
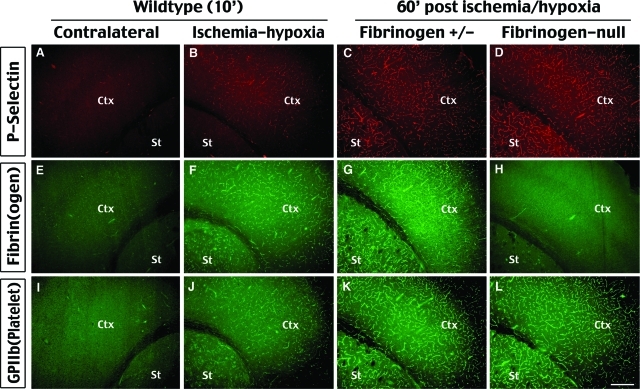
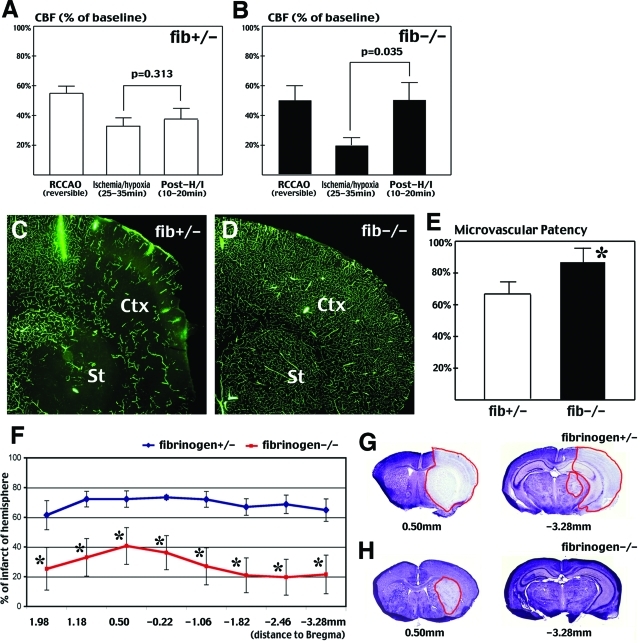
Hypoxia is a critical factor for cell death or survival in ischemic stroke, but the pathological consequences of combined ischemia-hypoxia are not fully understood. Here we examine this issue using a modified Levine/Vannucci procedure in adult mice that consists of unilateral common carotid artery occlusion and hypoxia with tightly regulated body temperature. At the cellular level, ischemia-hypoxia produced proinflammatory cytokines and simultaneously activated both prosurvival (eg, synthesis of heat shock 70 protein, phosphorylation of ERK and AKT) and proapoptosis signaling pathways (eg, release of cytochrome c and AIF from mitochondria, cleavage of caspase-9 and -8). However, caspase-3 was not activated, and very few cells completed the apoptosis process. Instead, many damaged neurons showed features of autophagic/lysosomal cell death. At the tissue level, ischemia-hypoxia caused persistent cerebral perfusion deficits even after release of the carotid artery occlusion. These changes were associated with both platelet deposition and fibrin accumulation within the cerebral circulation and would be expected to contribute to infarction. Complementary studies in fibrinogen-deficient mice revealed that the absence of fibrin and/or secondary fibrin-mediated inflammatory processes significantly attenuated brain damage. Together, these results suggest that ischemia-hypoxia is a powerful stimulus for spontaneous coagulation leading to reperfusion deficits and autophagic/lysosomal cell death in brain.
Figures
Comment in
-
The roles of autophagy in cerebral ischemia.Autophagy. 2007 Jan-Feb;3(1):42-4. doi: 10.4161/auto.3412. Epub 2007 Jan 18. Autophagy. 2007. PMID: 17035724 Review.
References
-
- Baron JC. Perfusion thresholds in human cerebral ischemia: historical perspective and therapeutic implications. Cerebrovasc Dis. 2001;11(Suppl 1):S2–S8. - PubMed
-
- Sobesky J, Zaro Weber O, Lehnhardt FG, Hesselmann V, Neveling M, Jacobs A, Heiss WD. Does the mismatch match the penumbra? Magnetic resonance imaging and positron emission tomography in early ischemic stroke. Stroke. 2005;36:980–985. - PubMed
-
- Yan SF, Mackman N, Kisiel W, Stern DM, Pinsky DJ. Hypoxia/hypoxemia-induced activation of the procoagulant pathways and the pathogenesis of ischemia-associated thrombosis. Arterioscler Thromb Vasc Biol. 1999;19:2029–2035. - PubMed
-
- Ten VS, Pinsky DJ. Endothelial response to hypoxia: physiologic adaptation and pathologic dysfunction. Curr Opin Crit Care. 2002;8:242–250. - PubMed
-
- Majno G, Ames AI, Chiang J, Wright R. No reflow after cerebral ischaemia. Lancet. 1967;2:569–570.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
