

Multiple-basin energy landscapes for large-amplitude conformational motions of proteins: Structure-based molecular dynamics simulations
- PMID: 16877541
- PMCID: PMC1567665
- DOI: 10.1073/pnas.0604375103
Multiple-basin energy landscapes for large-amplitude conformational motions of proteins: Structure-based molecular dynamics simulations
Abstract
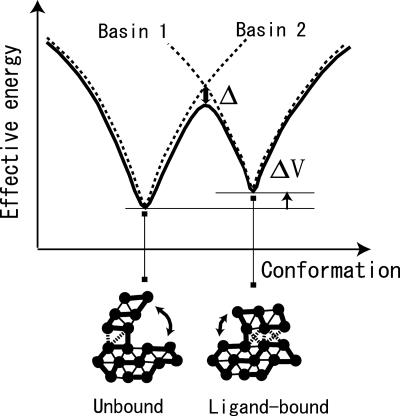
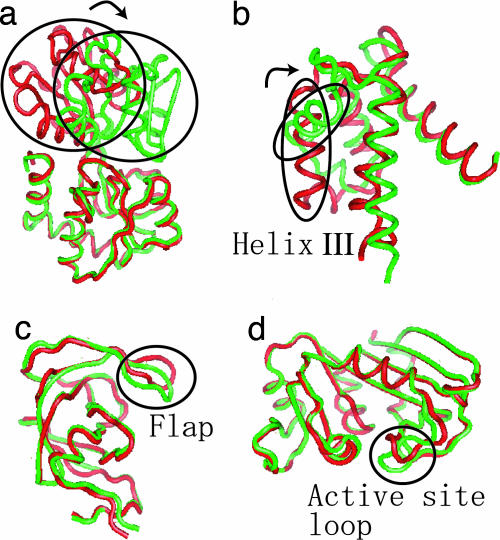
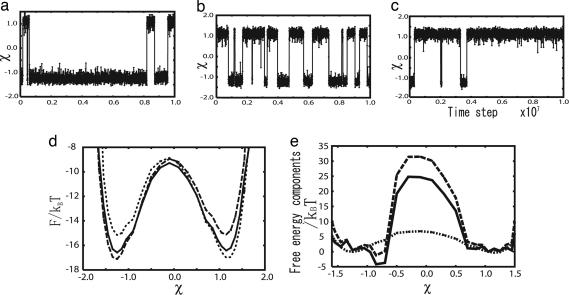
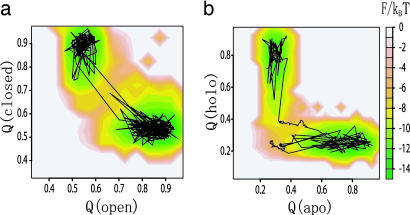
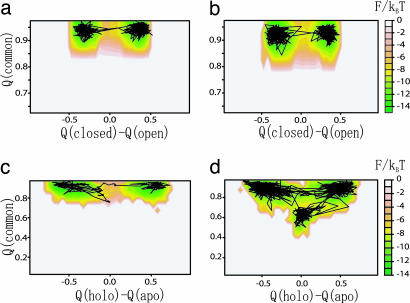
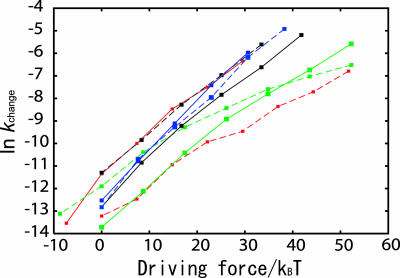
Biomolecules often undergo large-amplitude motions when they bind or release other molecules. Unlike macroscopic machines, these biomolecular machines can partially disassemble (unfold) and then reassemble (fold) during such transitions. Here we put forward a minimal structure-based model, the "multiple-basin model," that can directly be used for molecular dynamics simulation of even very large biomolecular systems so long as the endpoints of the conformational change are known. We investigate the model by simulating large-scale motions of four proteins: glutamine-binding protein, S100A6, dihydrofolate reductase, and HIV-1 protease. The mechanisms of conformational transition depend on the protein basin topologies and change with temperature near the folding transition. The conformational transition rate varies linearly with driving force over a fairly large range. This linearity appears to be a consequence of partial unfolding during the conformational transition.
Conflict of interest statement
Conflict of interest statement: No conflicts declared.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
