

Energetics of aliphatic deletions in protein cores
- PMID: 16877708
- PMCID: PMC2242576
- DOI: 10.1110/ps.062274906
Energetics of aliphatic deletions in protein cores
Abstract
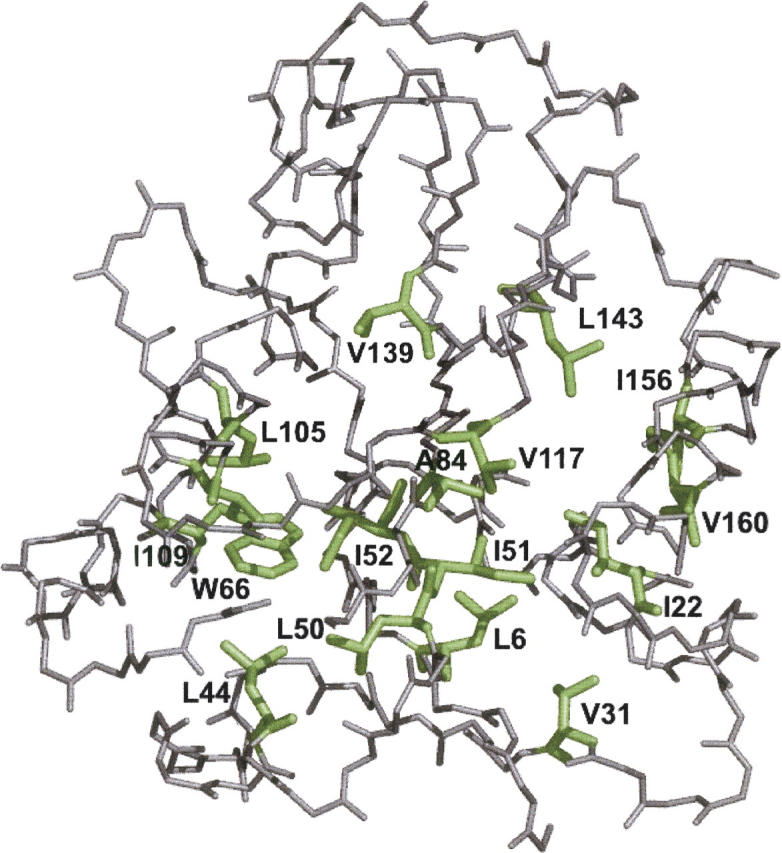
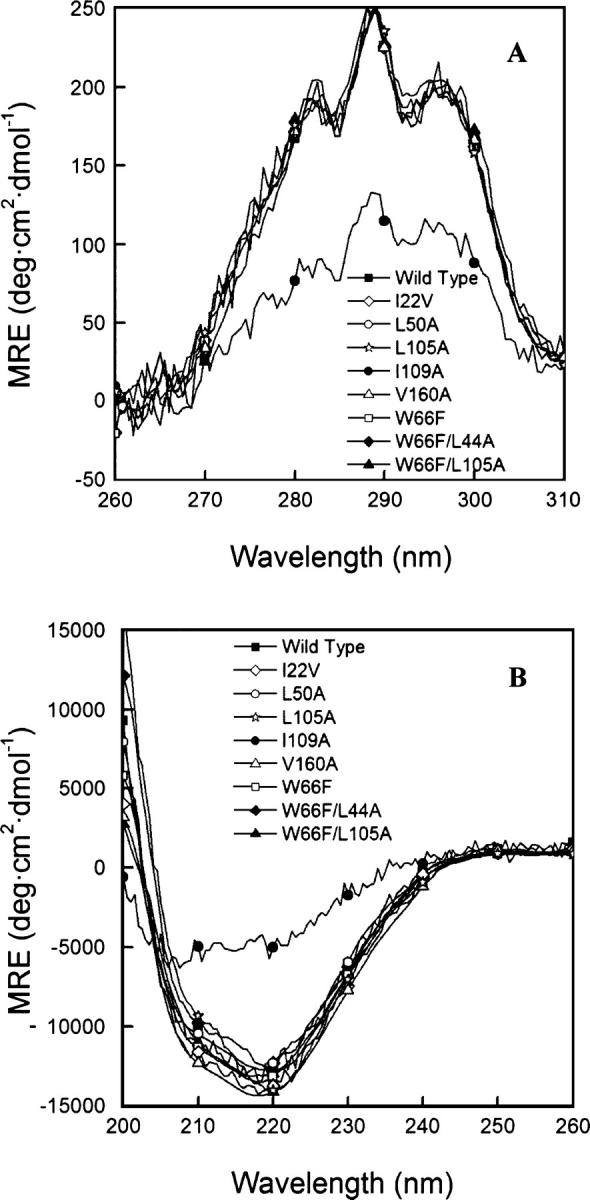
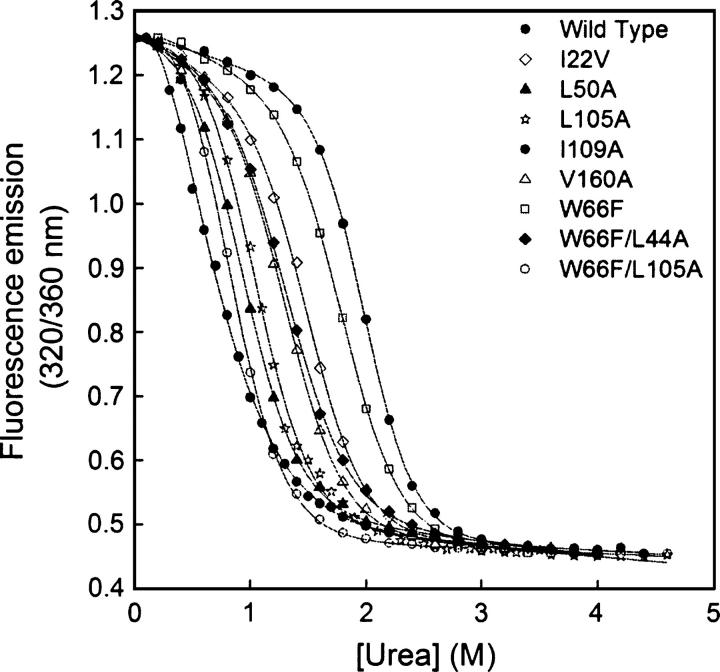
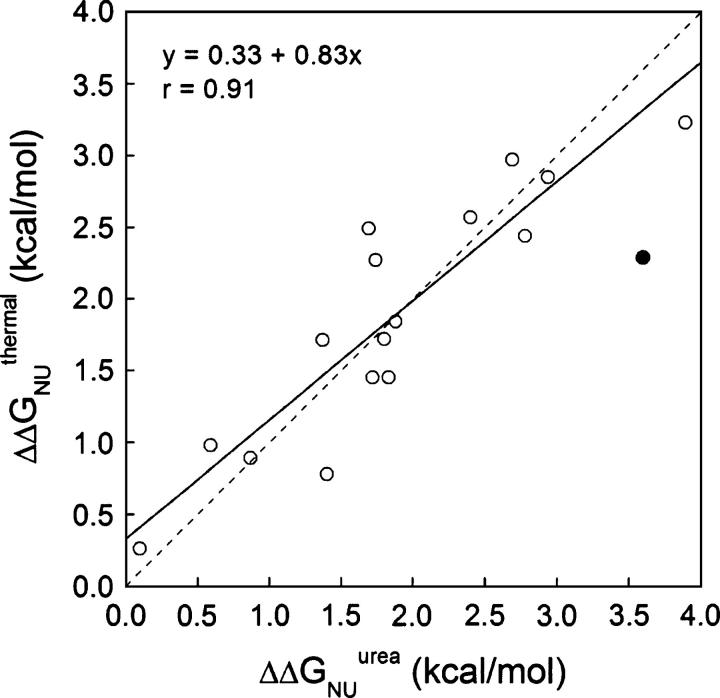
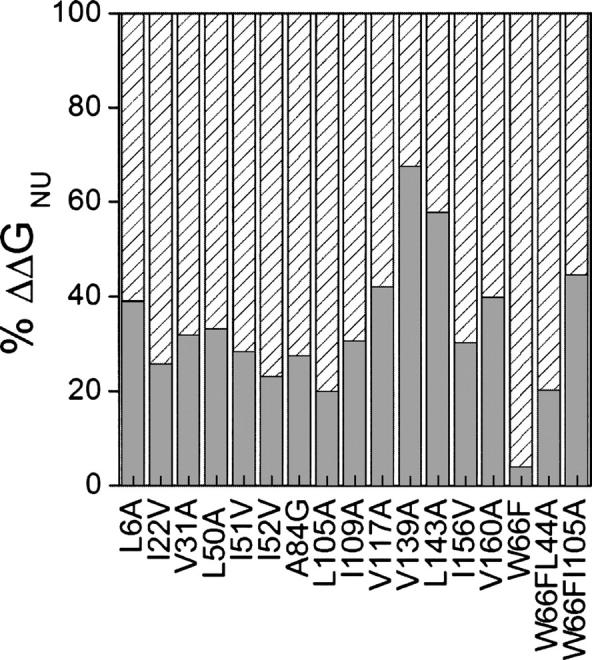
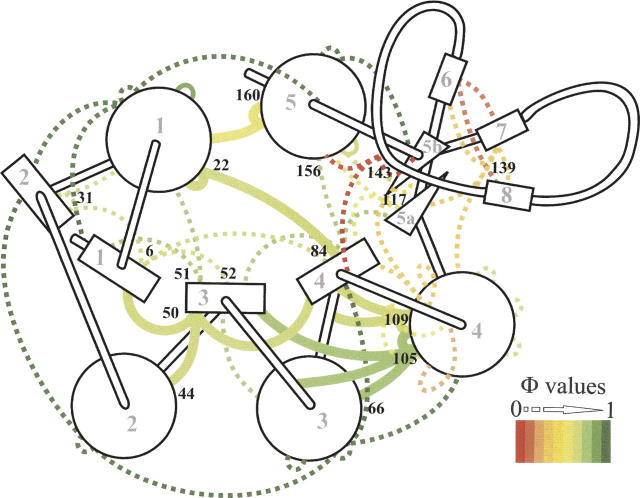
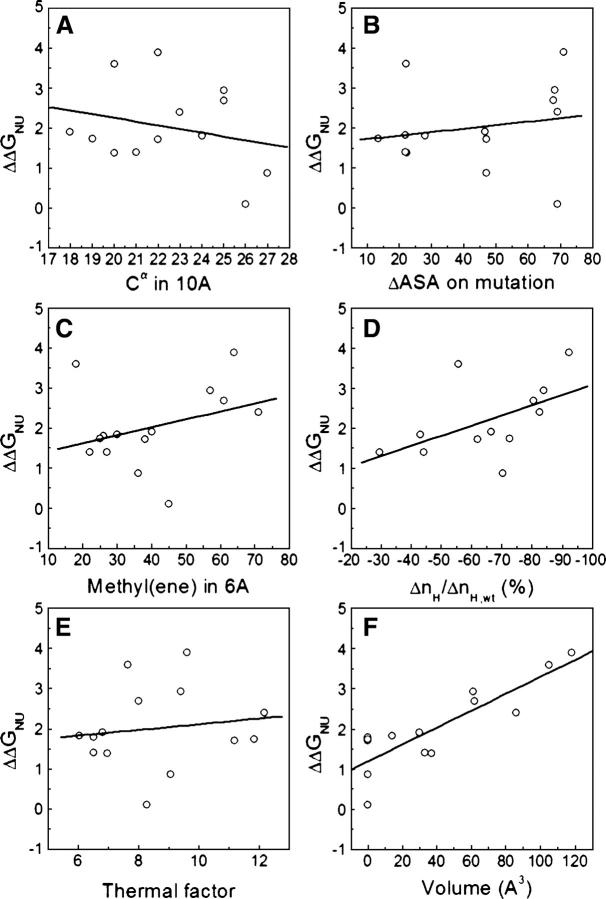
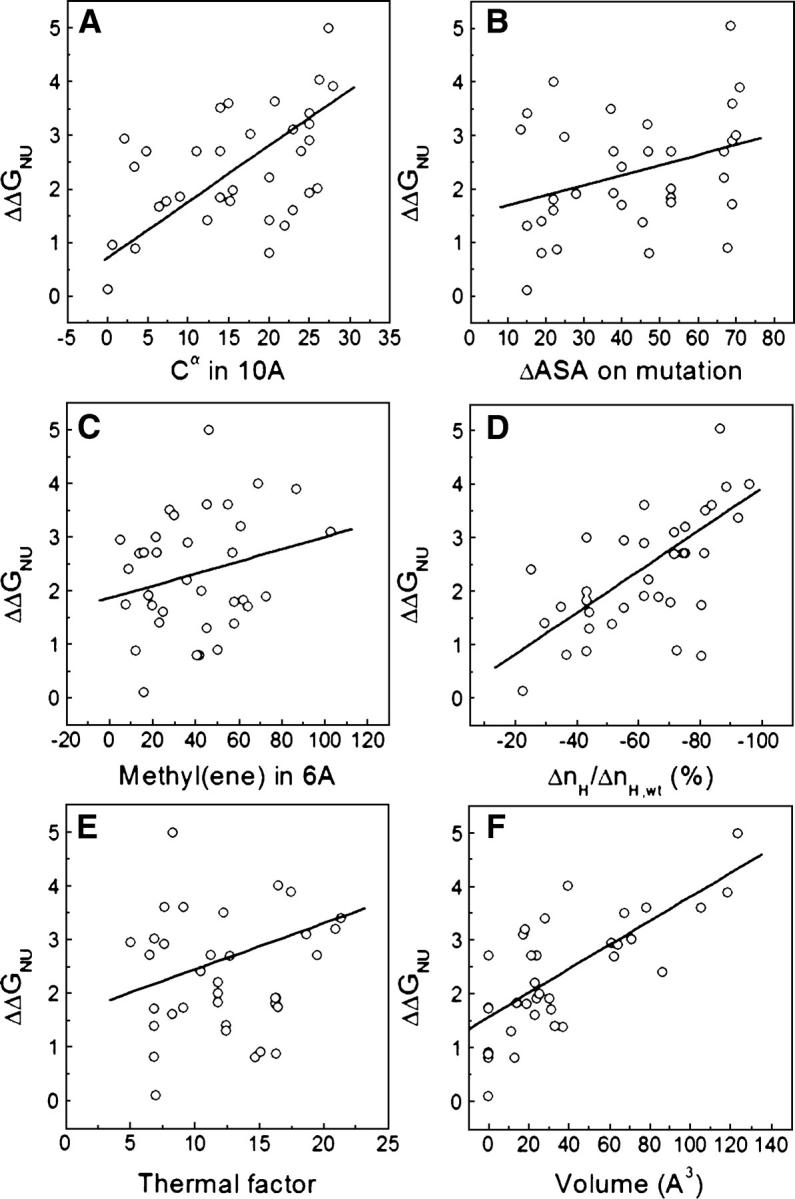
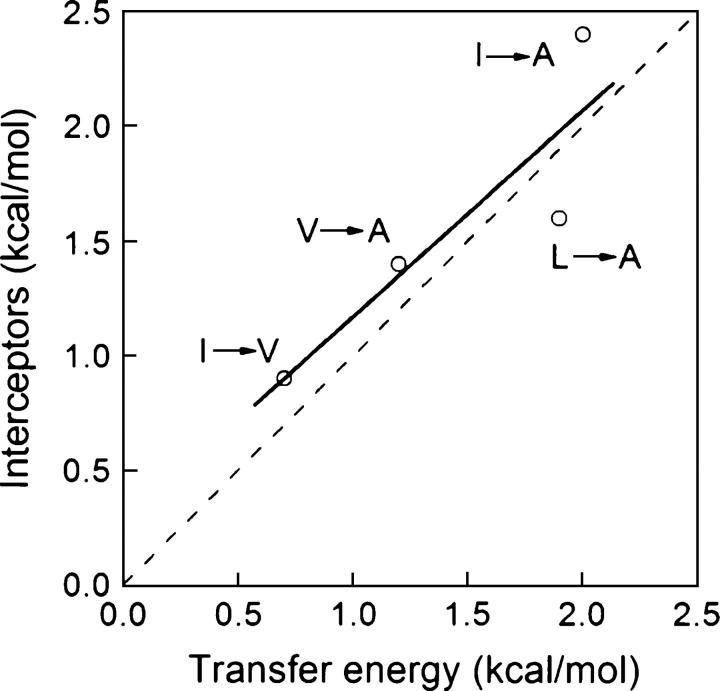
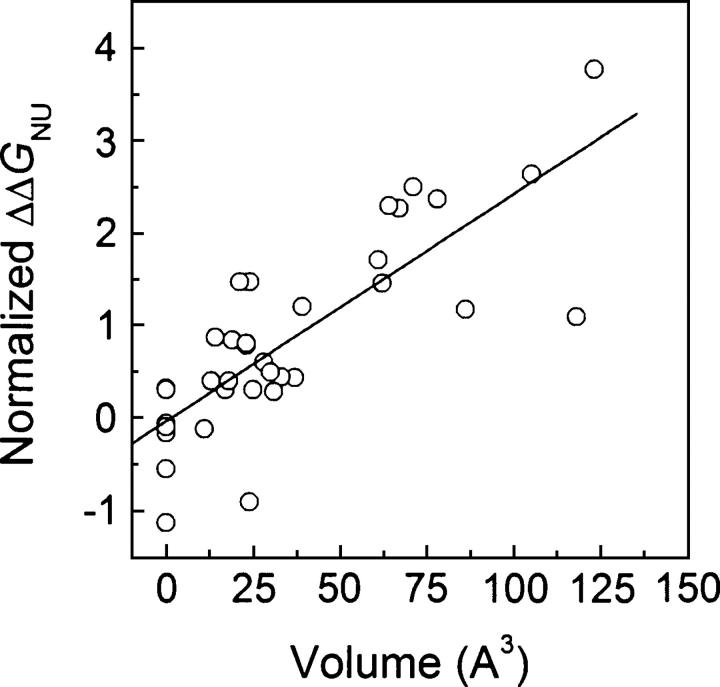
Although core residues can sometimes be replaced by shorter ones without introducing significant changes in protein structure, the energetic consequences are typically large and destabilizing. Many efforts have been devoted to understand and predict changes in stability from analysis of the environment of mutated residues, but the relationships proposed for individual proteins have often failed to describe additional data. We report here 17 apoflavodoxin large-to-small mutations that cause overall protein destabilizations of 0.6-3.9 kcal.mol(-1). By comparing two-state urea and three-state thermal unfolding data, the overall destabilizations observed are partitioned into effects on the N-to-I and on the I-to-U equilibria. In all cases, the equilibrium intermediate exerts a "buffering" effect that reduces the impact of the overall destabilization on the N-to-I equilibrium. The performance of several structure-energetics relationships, proposed to explain the energetics of hydrophobic shortening mutations, has been evaluated by using an apoflavodoxin data set consisting of 14 mutations involving branching-conservative aliphatic side-chain shortenings and a larger data set, including similar mutations implemented in seven model proteins. Our analysis shows that the stability changes observed for any of the different types of mutations (LA, IA, IV, and VA) in either data set are best explained by a combination of differential hydrophobicity and of the calculated volume of the modeled cavity (as previously observed for LA and IA mutations in lysozyme T4). In contrast, sequence conservation within the flavodoxin family, which is a good predictor for charge-reversal stabilizing mutations, does not perform so well for aliphatic shortening ones.
Figures
Similar articles
-
Filling small, empty protein cavities: structural and energetic consequences.J Mol Biol. 2006 May 5;358(3):701-12. doi: 10.1016/j.jmb.2006.02.060. Epub 2006 Mar 10. J Mol Biol. 2006. PMID: 16563433
-
Do proteins with similar folds have similar transition state structures? A diffuse transition state of the 169 residue apoflavodoxin.J Mol Biol. 2006 Jun 9;359(3):813-24. doi: 10.1016/j.jmb.2006.03.067. Epub 2006 Apr 19. J Mol Biol. 2006. PMID: 16647718
-
Do proteins always benefit from a stability increase? Relevant and residual stabilisation in a three-state protein by charge optimisation.J Mol Biol. 2004 Nov 12;344(1):223-37. doi: 10.1016/j.jmb.2004.09.047. J Mol Biol. 2004. PMID: 15504413
-
Native hydrogen bonds in a molten globule: the apoflavodoxin thermal intermediate.J Mol Biol. 2001 Mar 2;306(4):877-88. doi: 10.1006/jmbi.2001.4436. J Mol Biol. 2001. PMID: 11243795
-
Equilibrium phi-analysis of a molten globule: the 1-149 apoflavodoxin fragment.J Mol Biol. 2006 Feb 17;356(2):354-66. doi: 10.1016/j.jmb.2005.10.086. Epub 2005 Nov 28. J Mol Biol. 2006. PMID: 16364364
Cited by
-
Structural and energetic basis of carbohydrate-aromatic packing interactions in proteins.J Am Chem Soc. 2013 Jul 3;135(26):9877-84. doi: 10.1021/ja4040472. Epub 2013 Jun 19. J Am Chem Soc. 2013. PMID: 23742246 Free PMC article.
-
Interplay between drying and stability of a TIM barrel protein: a combined simulation-experimental study.J Am Chem Soc. 2013 Feb 6;135(5):1882-90. doi: 10.1021/ja310544t. Epub 2013 Jan 25. J Am Chem Soc. 2013. PMID: 23293932 Free PMC article.
-
A look at the face of the molten globule: Structural model of the Helicobacter pylori apoflavodoxin ensemble at acidic pH.Protein Sci. 2022 Nov;31(11):e4445. doi: 10.1002/pro.4445. Protein Sci. 2022. PMID: 36156320 Free PMC article.
-
Net Evolutionary Loss of Residue Polarity in Drosophilid Protein Cores Indicates Ongoing Optimization of Amino Acid Composition.Genome Biol Evol. 2017 Oct 1;9(10):2879-2892. doi: 10.1093/gbe/evx191. Genome Biol Evol. 2017. PMID: 28985302 Free PMC article.
-
Calculation of Protein Folding Thermodynamics Using Molecular Dynamics Simulations.J Chem Inf Model. 2023 Dec 25;63(24):7791-7806. doi: 10.1021/acs.jcim.3c01107. Epub 2023 Nov 13. J Chem Inf Model. 2023. PMID: 37955428 Free PMC article.
References
-
- Adamek D.H., Guerrero L., Blaber M., Caspar D.L. 2005. Structural and energetic consequences of mutations in a solvated hydrophobic cavity. J. Mol. Biol. 346: 307–318. - PubMed
-
- Akasako A., Haruki M., Oobatake M., Kanaya S. 1997. Conformational stabilities of Escherichia coli RNase HI variants with a series of amino acid substitutions at a cavity within the hydrophobic core. J. Biol. Chem. 272: 18686–18693. - PubMed
-
- Alber T., Sun D.P., Nye J.A., Muchmore D.C., Matthews B.W. 1987. Temperature-sensitive mutations of bacteriophage T4 lysozyme occur at sites with low mobility and low solvent accessibility in the folded protein. Biochemistry 26: 3754–3758. - PubMed
-
- Baase W.A., Eriksson A.E., Zhang X.J., Heinz D.W., Sauer U., Blaber M., Baldwin E.P., Wozniak J.A., Matthews B.W. 1992. Dissection of protein structure and folding by directed mutagenesis. Faraday Discuss. 93: 173–181. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
