

Advances and challenges in liquid chromatography-mass spectrometry-based proteomics profiling for clinical applications
- PMID: 16887931
- PMCID: PMC1781927
- DOI: 10.1074/mcp.M600162-MCP200
Advances and challenges in liquid chromatography-mass spectrometry-based proteomics profiling for clinical applications
Abstract
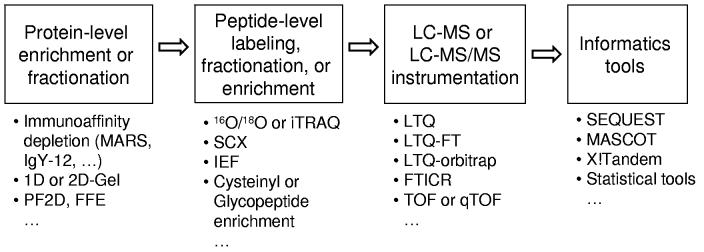
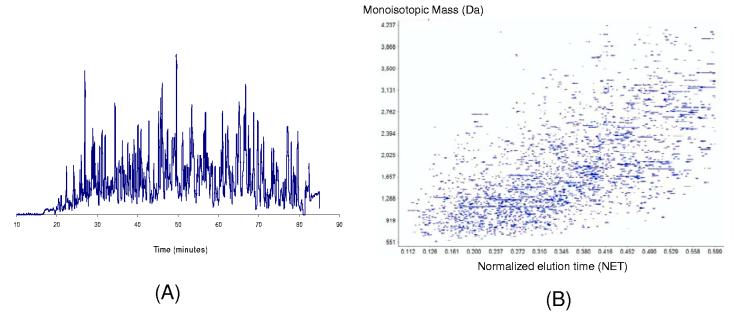
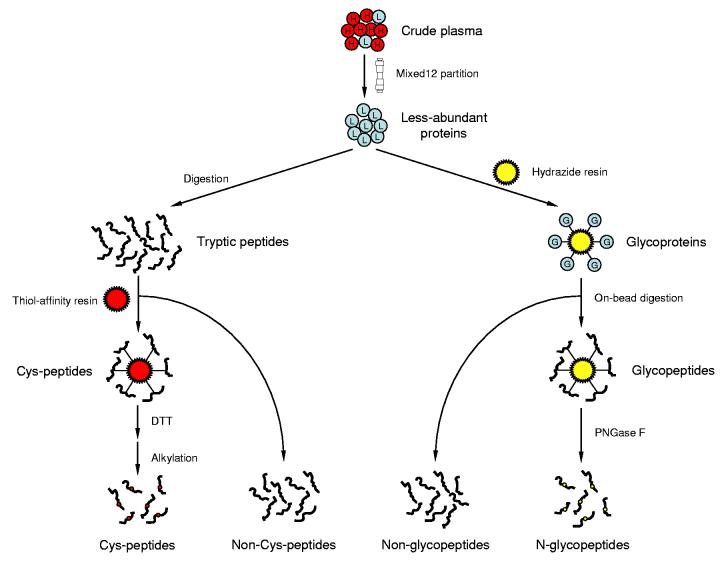
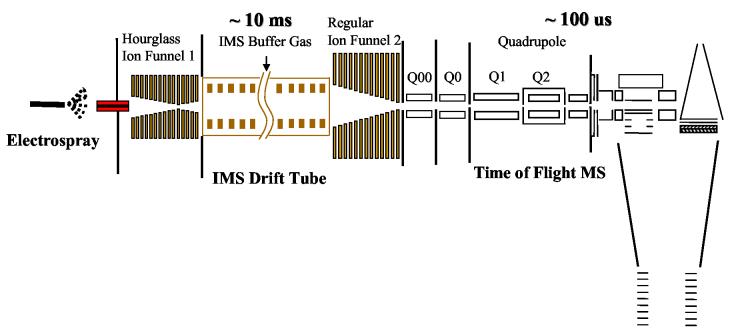
Recent advances in proteomics technologies provide tremendous opportunities for biomarker-related clinical applications; however, the distinctive characteristics of human biofluids such as the high dynamic range in protein abundances and extreme complexity of the proteomes present tremendous challenges. In this review we summarize recent advances in LC-MS-based proteomics profiling and its applications in clinical proteomics as well as discuss the major challenges associated with implementing these technologies for more effective candidate biomarker discovery. Developments in immunoaffinity depletion and various fractionation approaches in combination with substantial improvements in LC-MS platforms have enabled the plasma proteome to be profiled with considerably greater dynamic range of coverage, allowing many proteins at low ng/ml levels to be confidently identified. Despite these significant advances and efforts, major challenges associated with the dynamic range of measurements and extent of proteome coverage, confidence of peptide/protein identifications, quantitation accuracy, analysis throughput, and the robustness of present instrumentation must be addressed before a proteomics profiling platform suitable for efficient clinical applications can be routinely implemented.
Figures
References
-
- Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422:198–207. - PubMed
-
- Hanash S. Disease proteomics. Nature. 2003;422:226–232. - PubMed
-
- Etzioni R, Urban N, Ramsey S, McIntosh M, Schwartz S, Reid B, Radich J, Anderson G, Hartwell L. The case for early detection. Nat Rev Cancer. 2003;3:243–252. - PubMed
-
- Ludwig JA, Weinstein JN. Biomarkers in cancer staging, prognosis and treatment selection. Nat Rev Cancer. 2005;5:845–856. - PubMed
-
- Anderson NL, Anderson NG. The human plasma proteome: history, character, and diagnostic prospects. Mol. Cell. Proteomics. 2002;1:845–867. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
