

Analysis of microarray experiments of gene expression profiling
- PMID: 16890548
- PMCID: PMC2435252
- DOI: 10.1016/j.ajog.2006.07.001
Analysis of microarray experiments of gene expression profiling
Abstract
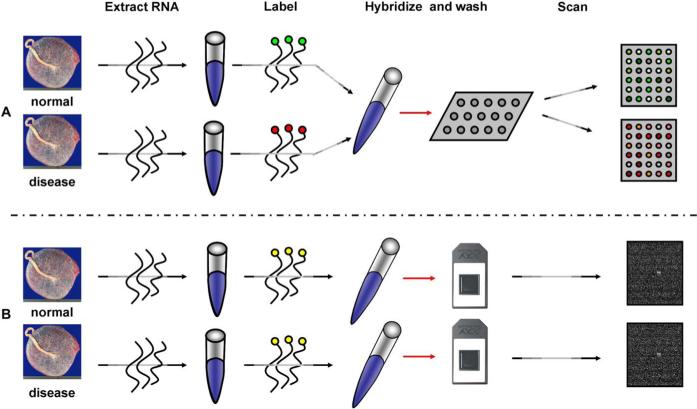
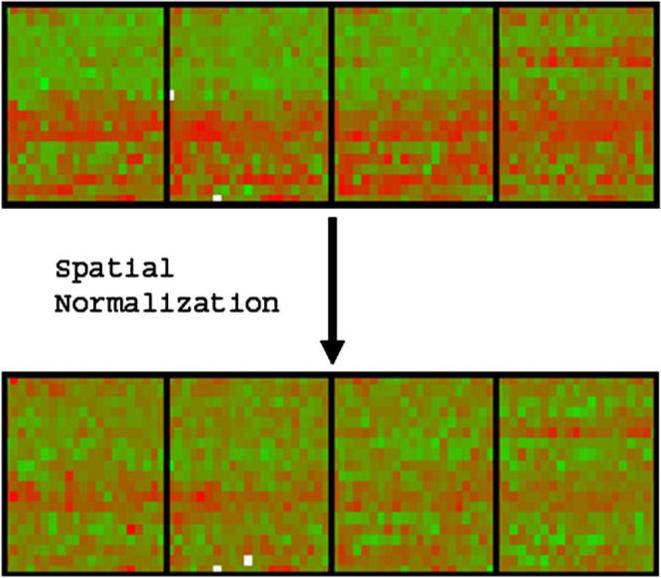
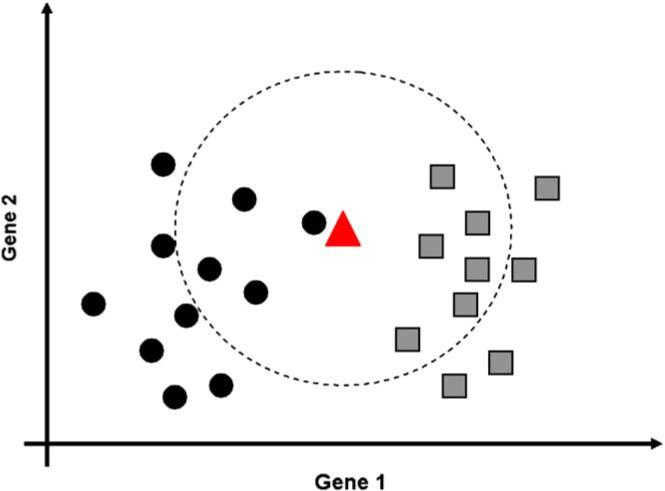
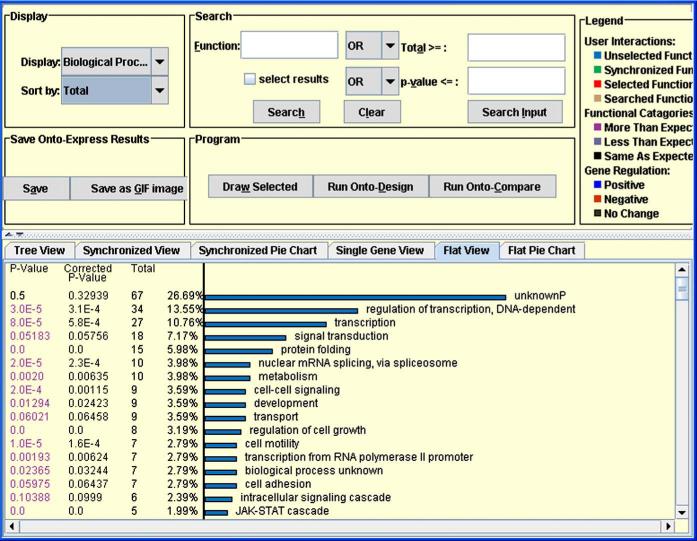
The study of gene expression profiling of cells and tissue has become a major tool for discovery in medicine. Microarray experiments allow description of genome-wide expression changes in health and disease. The results of such experiments are expected to change the methods employed in the diagnosis and prognosis of disease in obstetrics and gynecology. Moreover, an unbiased and systematic study of gene expression profiling should allow the establishment of a new taxonomy of disease for obstetric and gynecologic syndromes. Thus, a new era is emerging in which reproductive processes and disorders could be characterized using molecular tools and fingerprinting. The design, analysis, and interpretation of microarray experiments require specialized knowledge that is not part of the standard curriculum of our discipline. This article describes the types of studies that can be conducted with microarray experiments (class comparison, class prediction, class discovery). We discuss key issues pertaining to experimental design, data preprocessing, and gene selection methods. Common types of data representation are illustrated. Potential pitfalls in the interpretation of microarray experiments, as well as the strengths and limitations of this technology, are highlighted. This article is intended to assist clinicians in appraising the quality of the scientific evidence now reported in the obstetric and gynecologic literature.
Figures



Comment in
-
Commentary on microarray analysis for gynecologists and obstetricians.Am J Obstet Gynecol. 2007 Sep;197(3):330-1; author reply 331. doi: 10.1016/j.ajog.2007.05.025. Am J Obstet Gynecol. 2007. PMID: 17826449 No abstract available.
References
-
- Schena M, Shalon D, Davis RW, Brown PO. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science. 1995;270:467–70. - PubMed
-
- Schena M. Microarray biochip technology. Eaton Publishing; Sunnyvale, CA: 2000.
-
- Aguan K, Carvajal JA, Thompson LP, Weiner CP. Application of a functional genomics approach to identify differentially expressed genes in human myometrium during pregnancy and labour. Mol Hum Reprod. 2000;6:1141–5. - PubMed
-
- Berchuck A, Iversen ES, Lancaster JM, Dressman HK, West M, Nevins JR, et al. Prediction of optimal versus suboptimal cytoreduction of advanced-stage serous ovarian cancer with the use of microarrays. Am J Obstet Gynecol. 2004;190:910–25. - PubMed
-
- Bethin KE, Nagai Y, Sladek R, Asada M, Sadovsky Y, Hudson TJ, et al. Microarray analysis of uterine gene expression in mouse and human pregnancy. Mol Endocrinol. 2003;17:1454–69. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
