

A new paradigm for DNA polymerase specificity
- PMID: 16893169
- PMCID: PMC7526746
- DOI: 10.1021/bi060993z
A new paradigm for DNA polymerase specificity
Abstract
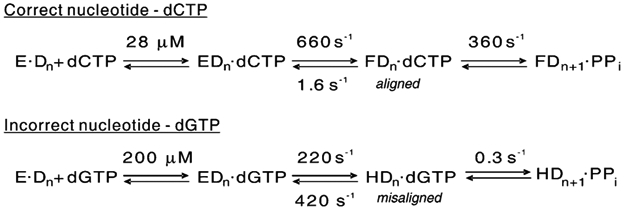
We show that T7 DNA polymerase exists in three distinct structural states, as reported by a conformationally sensitive fluorophore attached to the recognition (fingers) domain. The conformational change induced by a correct nucleotide commits the substrate to the forward reaction, and the slow reversal of the conformational change eliminates the rate of the chemistry step from any contribution toward enzyme specificity. Discrimination against mismatches is enhanced by the rapid release of mismatched nucleotides from the ternary E.DNA.deoxynucleoside triphosphate complex and by the use of substrate-binding energy to actively misalign catalytic residues to reduce the rate of misincorporation. Our refined model for enzyme selectivity extends traditional thermodynamic formalism by including substrate-induced structural alignment or misalignment of catalytic residues as a third dimension on the free-energy profile and by including the rate of substrate dissociation as a key kinetic parameter.
Figures
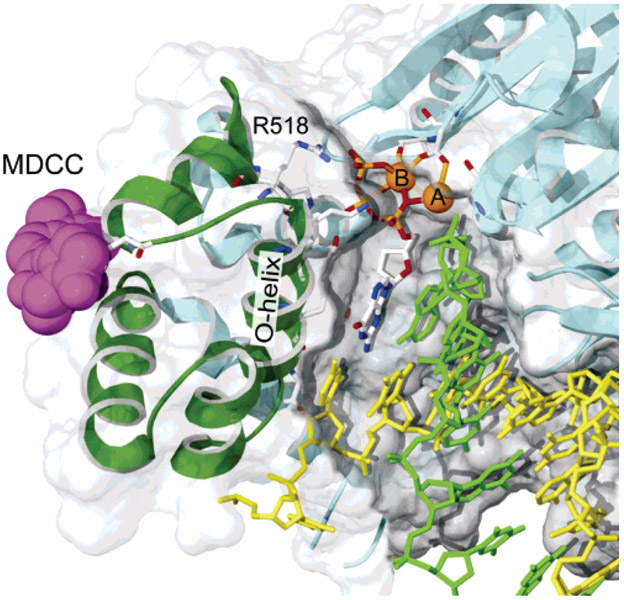

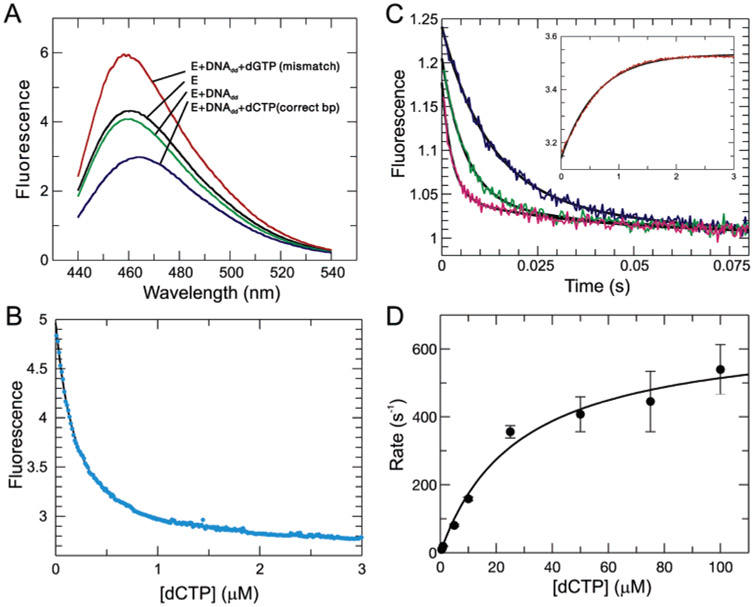
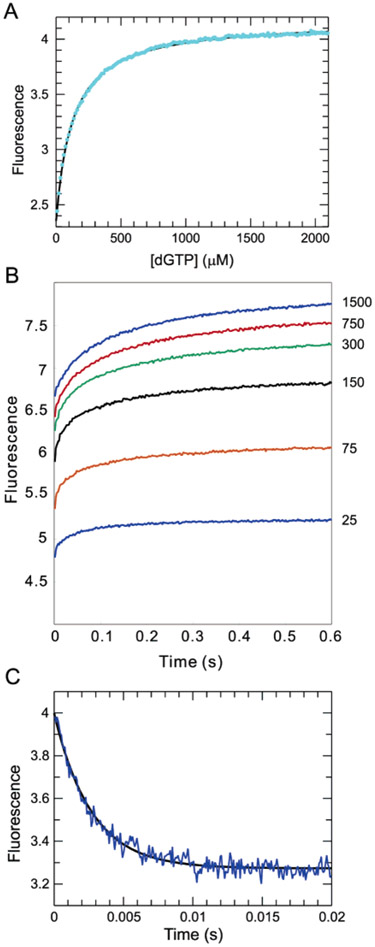
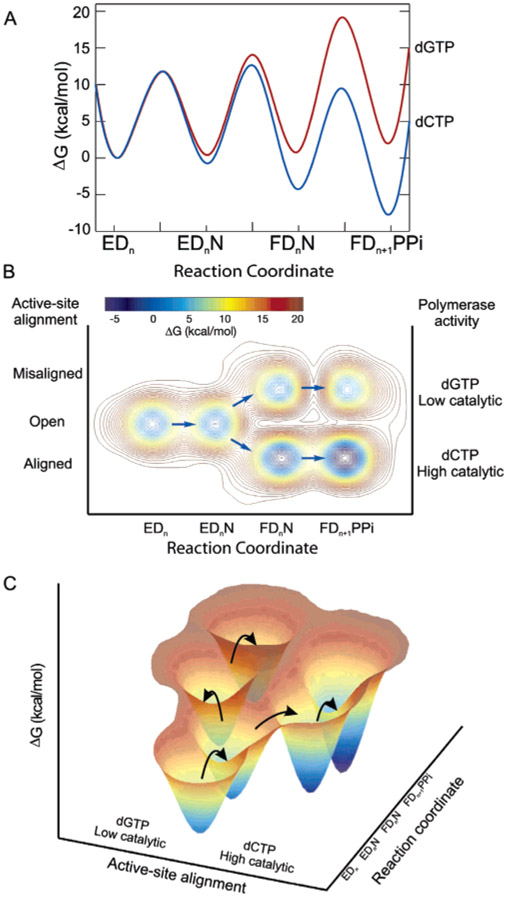
References
-
- Fersht A (1998) Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding, W. H. Freeman and Company, New York.
-
- Fersht A (1985) Enzyme Structure and Mechanism, W. H. Freeman and Company, New York.
-
- Fersht AR (1974) Catalysis, binding and enzyme-substrate complementarity, Proc. R. Soc. London, Ser. B 187, 397–407. - PubMed
-
- Post CB, and Ray WJ Jr. (1995) Re-examination of induced fit as a determinant of substrate specificity in enzymatic reactions, Biochemistry 34, 15881–15885. - PubMed
-
- Johnson KA (1993) Conformational coupling in DNA polymerase fidelity, Annu. Rev. Biochem 62, 685–713. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
