

Graemlin: general and robust alignment of multiple large interaction networks
- PMID: 16899655
- PMCID: PMC1557769
- DOI: 10.1101/gr.5235706
Graemlin: general and robust alignment of multiple large interaction networks
Abstract
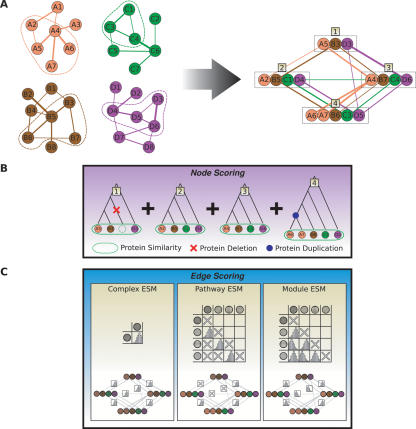
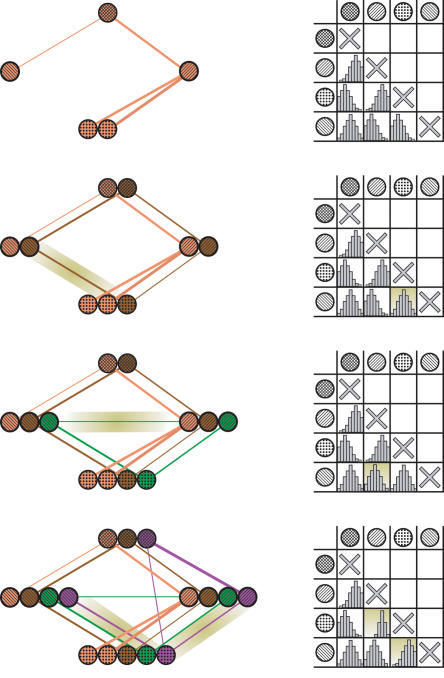
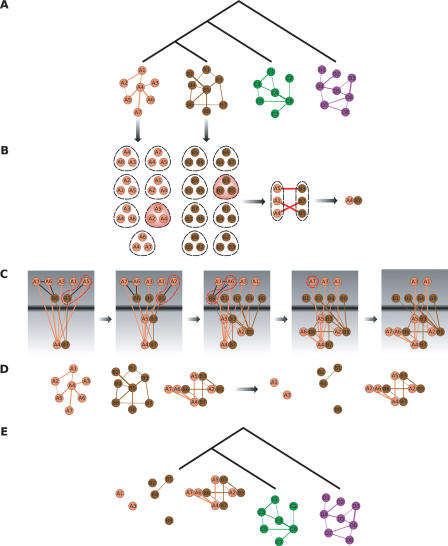
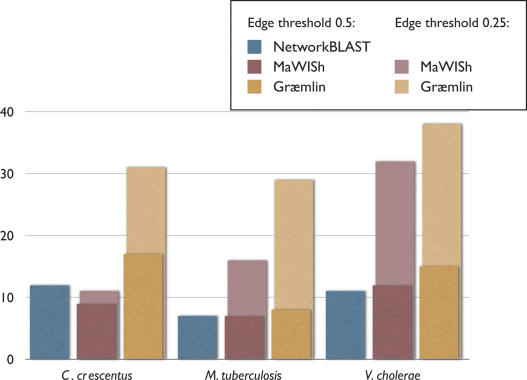
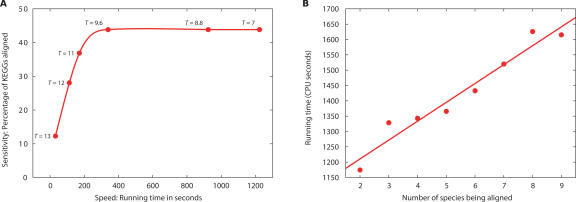
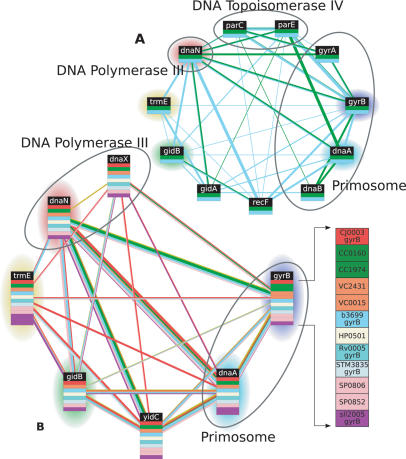
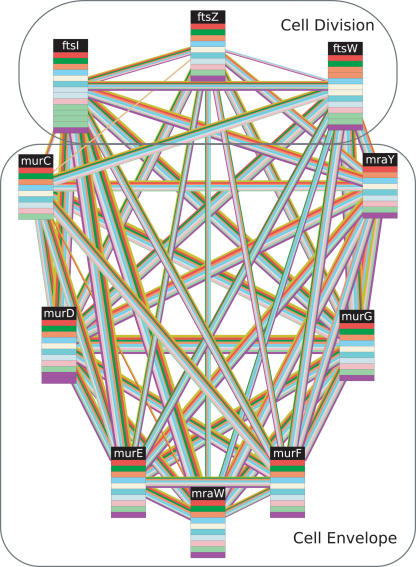
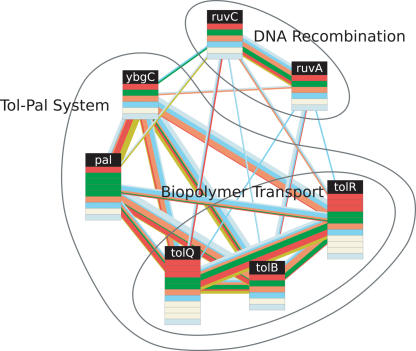
The recent proliferation of protein interaction networks has motivated research into network alignment: the cross-species comparison of conserved functional modules. Previous studies have laid the foundations for such comparisons and demonstrated their power on a select set of sparse interaction networks. Recently, however, new computational techniques have produced hundreds of predicted interaction networks with interconnection densities that push existing alignment algorithms to their limits. To find conserved functional modules in these new networks, we have developed Graemlin, the first algorithm capable of scalable multiple network alignment. Graemlin's explicit model of functional evolution allows both the generalization of existing alignment scoring schemes and the location of conserved network topologies other than protein complexes and metabolic pathways. To assess Graemlin's performance, we have developed the first quantitative benchmarks for network alignment, which allow comparisons of algorithms in terms of their ability to recapitulate the KEGG database of conserved functional modules. We find that Graemlin achieves substantial scalability gains over previous methods while improving sensitivity.
Figures
Similar articles
-
LocalAli: an evolutionary-based local alignment approach to identify functionally conserved modules in multiple networks.Bioinformatics. 2015 Feb 1;31(3):363-72. doi: 10.1093/bioinformatics/btu652. Epub 2014 Oct 4. Bioinformatics. 2015. PMID: 25282642
-
LePrimAlign: local entropy-based alignment of PPI networks to predict conserved modules.BMC Genomics. 2019 Dec 24;20(Suppl 9):964. doi: 10.1186/s12864-019-6271-3. BMC Genomics. 2019. PMID: 31874635 Free PMC article.
-
Global alignment of protein-protein interaction networks.Methods Mol Biol. 2013;939:21-34. doi: 10.1007/978-1-62703-107-3_3. Methods Mol Biol. 2013. PMID: 23192538
-
Computational reconstruction of protein-protein interaction networks: algorithms and issues.Methods Mol Biol. 2009;541:89-100. doi: 10.1007/978-1-59745-243-4_5. Methods Mol Biol. 2009. PMID: 19381528 Review.
-
Identifying protein complexes and functional modules--from static PPI networks to dynamic PPI networks.Brief Bioinform. 2014 Mar;15(2):177-94. doi: 10.1093/bib/bbt039. Epub 2013 Jun 18. Brief Bioinform. 2014. PMID: 23780996 Review.
Cited by
-
GreedyPlus: An Algorithm for the Alignment of Interface Interaction Networks.Sci Rep. 2015 Jul 13;5:12074. doi: 10.1038/srep12074. Sci Rep. 2015. PMID: 26165520 Free PMC article.
-
From homogeneous to heterogeneous network alignment via colored graphlets.Sci Rep. 2018 Aug 21;8(1):12524. doi: 10.1038/s41598-018-30831-w. Sci Rep. 2018. PMID: 30131590 Free PMC article.
-
Identification of co-evolving temporal networks.BMC Genomics. 2019 Jun 13;20(Suppl 6):434. doi: 10.1186/s12864-019-5719-9. BMC Genomics. 2019. PMID: 31189471 Free PMC article.
-
Pairwise Biological Network Alignment Based on Discrete Bat Algorithm.Comput Math Methods Med. 2021 Nov 3;2021:5548993. doi: 10.1155/2021/5548993. eCollection 2021. Comput Math Methods Med. 2021. PMID: 34777564 Free PMC article.
-
Inferring strain-level mutational drivers of phage-bacteria interaction phenotypes arising during coevolutionary dynamics.bioRxiv [Preprint]. 2024 Nov 21:2024.01.08.574707. doi: 10.1101/2024.01.08.574707. bioRxiv. 2024. Update in: Virus Evol. 2024 Nov 29;10(1):veae104. doi: 10.1093/ve/veae104. PMID: 38260415 Free PMC article. Updated. Preprint.
References
-
- Altschul S.F., Carroll R.J., Lipman D.J., Carroll R.J., Lipman D.J., Lipman D.J. Weights for data related by a tree. J. Mol. Biol. 1989;207:647–653. - PubMed
-
- Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J., Gish W., Miller W., Myers E.W., Lipman D.J., Miller W., Myers E.W., Lipman D.J., Myers E.W., Lipman D.J., Lipman D.J. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. - PubMed
-
- Altschul S.F., Madden T.L., Schaffer A.A., Zhang J., Zhang Z., Miller W., Lipman D.J., Madden T.L., Schaffer A.A., Zhang J., Zhang Z., Miller W., Lipman D.J., Schaffer A.A., Zhang J., Zhang Z., Miller W., Lipman D.J., Zhang J., Zhang Z., Miller W., Lipman D.J., Zhang Z., Miller W., Lipman D.J., Miller W., Lipman D.J., Lipman D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. - PMC - PubMed
-
- Andreeva A., Howorth D., Brenner S.E., Hubbard T.J.P., Chothia C., Murzin A.G., Howorth D., Brenner S.E., Hubbard T.J.P., Chothia C., Murzin A.G., Brenner S.E., Hubbard T.J.P., Chothia C., Murzin A.G., Hubbard T.J.P., Chothia C., Murzin A.G., Chothia C., Murzin A.G., Murzin A.G. SCOP database in 2004: Refinements integrate structure and sequence family data. Nucleic Acids Res. 2004;32:D226–D229. - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources