

Correlation between clinical severity in patients with Rett syndrome with a p.R168X or p.T158M MECP2 mutation, and the direction and degree of skewing of X-chromosome inactivation
- PMID: 16905679
- PMCID: PMC2598067
- DOI: 10.1136/jmg.2006.045260
Correlation between clinical severity in patients with Rett syndrome with a p.R168X or p.T158M MECP2 mutation, and the direction and degree of skewing of X-chromosome inactivation
Abstract
Introduction: Rett syndrome (RTT) is an X-linked dominant neurodevelopmental disorder that is usually associated with mutations in the MECP2 gene. The most common mutations in the gene are p.R168X and p.T158M. The influence of X-chromosome inactivation (XCI) on clinical severity in patients with RTT with these mutations was investigated, taking into account the extent and direction of skewing.
Methods: Female patients and their parents were recruited from the UK and Australia. Clinical severity was measured by the Pineda Severity and Kerr profile scores. The degree of XCI and its direction relative to the X chromosome parent of origin were measured in DNA prepared from peripheral blood leucocytes, and allele-specific polymerase chain reaction was used to determine the parental origin of mutation. Combining these, the percentage of cells expected to express the mutant allele was calculated.
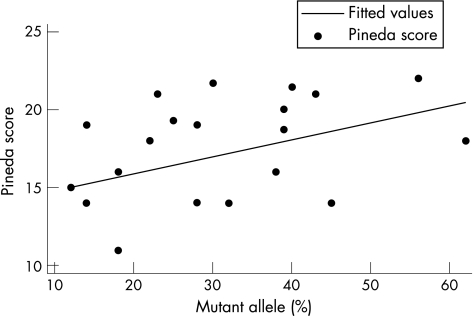
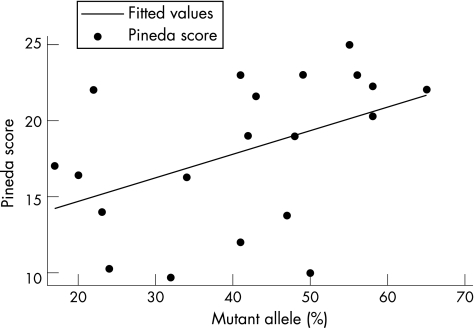
Results: Linear regression analysis was undertaken for fully informative cases with p.R168X (n = 23) and p.T158M (n = 20) mutations. A statistically significant increase in clinical severity with increase in the proportion of active mutated allele was shown for both the p.R168X and p.T158M mutations.
Conclusions: XCI may vary in neurological and haematological tissues. However, these data are the first to show a relationship between the degree and direction of XCI in leucocytes and clinical severity in RTT, although the clinical utility of this in giving a prognosis for individual patients is unclear.
Conflict of interest statement
Competing interests: None declared.
References
-
- Amir R E, Van dV I, Wan M, Tran C Q, Francke U, Zoghbi H Y. Rett syndrome is caused by mutations in X‐linked MECP2, encoding methyl‐CpG‐binding protein 2. Nat Genet 199923185–188. - PubMed
-
- Charman T, Neilson T C, Mash V, Archer H, Gardiner M T, Knudsen G P, McDonnell A, Perry J, Whatley S D, Bunyan D J, Ravn K, Mount R H, Hastings R P, Hulten M, Orstavik K H, Reilly S, Cass H, Clarke A, Kerr A M, Bailey M E S. Dimensional phenotypic analysis and functional categorisation of mutations reveal novel genotype‐phenotype associations in Rett syndrome. Eur J Hum Genet 2005131121–1130. - PubMed
-
- Ham A L, Kumar A, Deeter R, Schanen N C. Does genotype predict phenotype in Rett syndrome? J Child Neurol 200520768–778. - PubMed
-
- Gartler S M, Dyer K A, Graves J A, Rocchi M. A two step model for mammalian X‐chromosome inactivation. Prog Clin Biol Res 1985198223–235. - PubMed
-
- Lyon M F. X‐chromosome inactivation as a system of gene dosage compensation to regulate gene expression. Prog Nucleic Acid Res Mol Biol 198936119–130. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical