

A new autosomal recessive form of Stickler syndrome is caused by a mutation in the COL9A1 gene
- PMID: 16909383
- PMCID: PMC1559536
- DOI: 10.1086/506478
A new autosomal recessive form of Stickler syndrome is caused by a mutation in the COL9A1 gene
Abstract
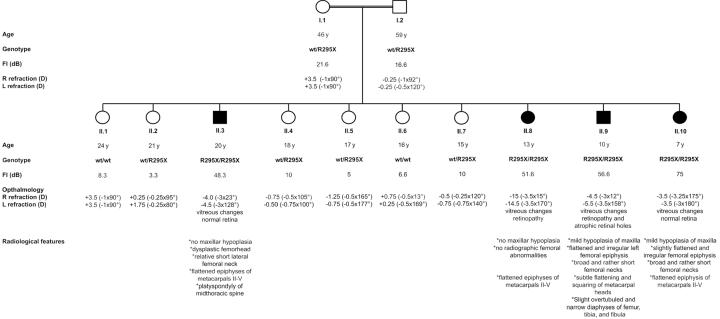
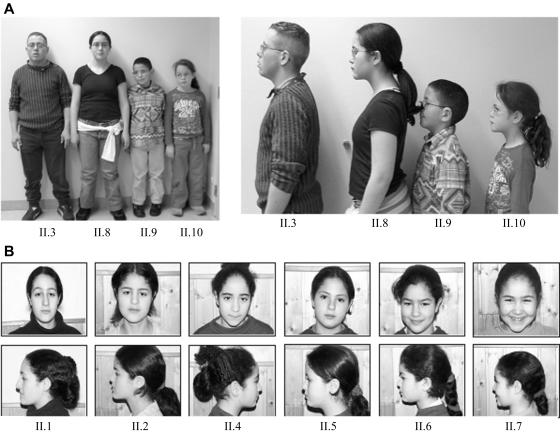
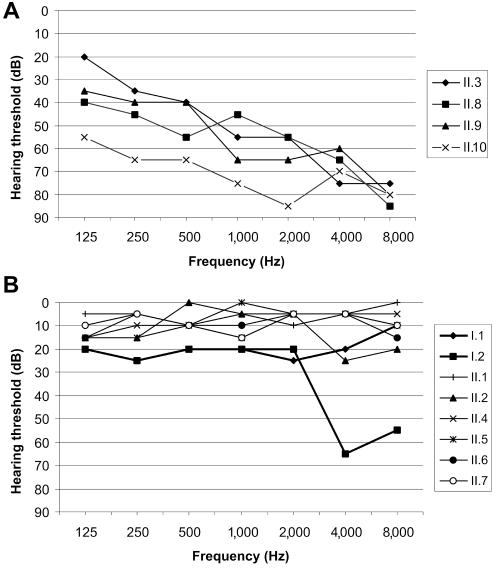
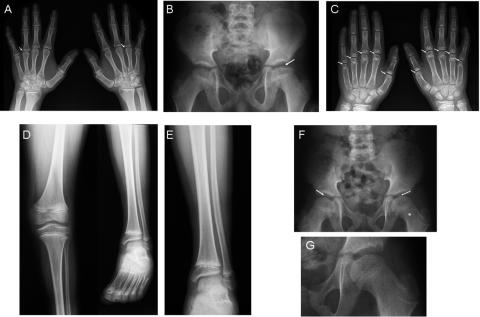
Stickler syndrome is characterized by ophthalmic, articular, orofacial, and auditory manifestations. It has an autosomal dominant inheritance pattern and is caused by mutations in COL2A1, COL11A1, and COL11A2. We describe a family of Moroccan origin that consists of four children with Stickler syndrome, six unaffected children, and two unaffected parents who are distant relatives (fifth degree). All family members were clinically investigated for ear, nose, and throat; ophthalmologic; and radiological abnormalities. Four children showed symptoms characteristic of Stickler syndrome, including moderate-to-severe sensorineural hearing loss, moderate-to-high myopia with vitreoretinopathy, and epiphyseal dysplasia. We considered the COL9A1 gene, located on chromosome 6q13, to be a candidate gene on the basis of the structural association with collagen types II and XI and because of the high expression in the human inner ear indicated by cDNA microarray. Mutation analysis of the coding region of the COL9A1 gene showed a homozygous R295X mutation in the four affected children. The parents and four unaffected children were heterozygous carriers of the R295X mutation. Two unaffected children were homozygous for the wild-type allele. None of the family members except the homozygous R295X carriers had any signs of Stickler syndrome. Therefore, COL9A1 is the fourth identified gene that can cause Stickler syndrome. In contrast to the three previously reported Stickler syndrome-causing genes, this gene causes a form of Stickler syndrome with an autosomal recessive inheritance pattern. This finding will have a major impact on the genetic counseling of patients with Stickler syndrome and on the understanding of the pathophysiology of collagens. Mutation analysis of this gene is recommended in patients with Stickler syndrome with possible autosomal recessive inheritance.
Figures
References
Web Resource
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/OMIM (for Stickler syndrome types I, II, and III)
References
-
- Stickler GB, Belau PG, Farrell FJ, Jones JD, Pugh DG, Steinberg AG, Ward LE (1965) Hereditary progressive arthro-ophthalmopathy. Mayo Clin Proc 40:433–455 - PubMed
-
- Williams CJ, Ganguly A, Considine E, McCarron S, Prockop DJ, Walsh-Vockley C, Michels VV (1996) A-2→G transition at the 3′ acceptor splice site of IVS17 characterizes the COL2A1 gene mutation in the original Stickler syndrome kindred. Am J Med Genet 63:461–46710.1002/(SICI)1096-8628(19960614)63:3<461::AID-AJMG9>3.0.CO;2-U - DOI - PubMed
-
- Vikkula M, Mariman EC, Lui VC, Zhidkova NI, Tiller GE, Goldring MB, van Beersum SE, de Waal Malefijt MC, van den Hoogen FHJ, Ropers H-H, Mayne R, Cheah KSE, Olsen BR, Warman ML, Bruuner HG (1995) Autosomal dominant and recessive osteochondrodysplasias associated with the COL11A2 locus. Cell 80:431–43710.1016/0092-8674(95)90493-X - DOI - PubMed
-
- Snead MP, Payne SJ, Barton DE, Yates JR, al-Imara L, Pope FM, Scott JD (1994) Stickler syndrome: correlation between vitreoretinal phenotypes and linkage to COL2A1. Eye 8:609–614 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
