

The value of molecular haplotypes in a family-based linkage study
- PMID: 16909384
- PMCID: PMC1559540
- DOI: 10.1086/506626
The value of molecular haplotypes in a family-based linkage study
Abstract
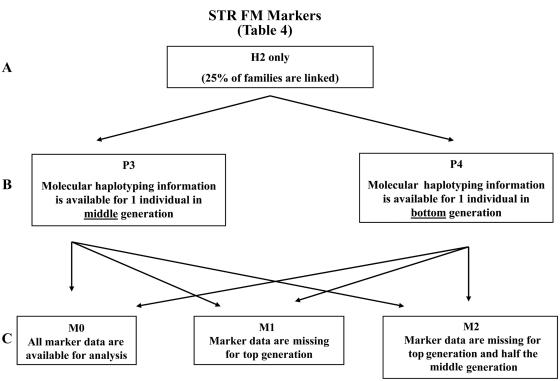
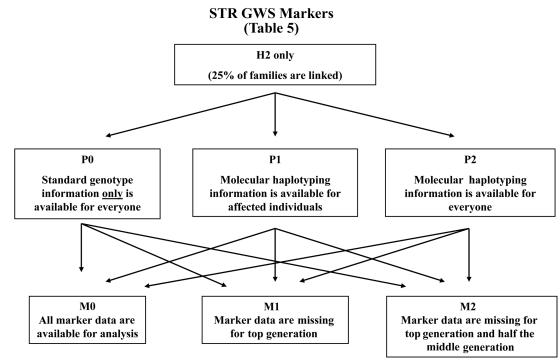
Novel methods that could improve the power of conventional methods of gene discovery for complex diseases should be investigated. In a simulation study, we aimed to investigate the value of molecular haplotypes in the context of a family-based linkage study. The term "haplotype" (or "haploid genotype") refers to syntenic alleles inherited on a single chromosome, and we use the term "molecular haplotype" to refer to haplotypes that have been determined directly by use of a molecular technique such as long-range allele-specific polymerase chain reaction. In our study, we simulated genotype and phenotype data and then compared the powers of analyzing these data under the assumptions that various levels of information from molecular haplotypes were available. (This information was available because of the simulation procedure.) Several conclusions can be drawn. First, as expected, when genetic homogeneity is expected or when marker data are complete, it is not efficient to generate molecular haplotyping information. However, with levels of heterogeneity and missing data patterns typical of complex diseases, we observed a 23%-77% relative increase in the power to detect linkage in the presence of heterogeneity with heterogeneity LOD scores >3.0 when all individuals are molecularly haplotyped (compared with the power when only standard genotypes are used). Furthermore, our simulations indicate that most of the increase in power can be achieved by molecularly haplotyping a single individual in each family, thereby making molecular haplotyping a valuable strategy for increasing the power of gene mapping studies of complex diseases. Maximization of power, given an existing family set, can be particularly important for late-onset, often-fatal diseases such as cancer, for which informative families are difficult to collect.
Figures

References
-
- Puffenberger EG, Kauffman ER, Bolk S, Matise TC, Washington SS, Angrist M, Weissenbach J, Garver KL, Mascari M, Ladda R, Siaugenhaupt S, Chakravarti A (1994) Identity-by-descent and association mapping of a recessive gene for Hirschsprung disease on human chromosome 13q22. Hum Mol Genet 3:1217–1225 - PubMed
-
- Drysdale CM, McGraw DW, Stack CB, Stephens JC, Judson RS, Nandabalan K, Arnold K, Ruano G, Liggett SB (2000) Complex promoter and coding region β 2-adrenergic receptor haplotypes alter receptor expression and predict in vivo responsiveness. Proc Natl Acad Sci USA 97:10483–1048810.1073/pnas.97.19.10483 - DOI - PMC - PubMed
-
- Hugot JP, Chamaillard M, Zouali H, Lesage S, Cezard JP, Belaiche J, Almer S, Tysk C, O’Morain CA, Gassull M, Binder V, Finkel Y, Cortot A, Modigliani R, Laurent-Puig P, Gower-Rousseau C, Macry J, Colombel JF, Sahbatou M, Thomas G (2001) Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature 411:599–60310.1038/35079107 - DOI - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
