

The mitochondrial fission protein hFis1 requires the endoplasmic reticulum gateway to induce apoptosis
- PMID: 16914522
- PMCID: PMC1635393
- DOI: 10.1091/mbc.e06-05-0377
The mitochondrial fission protein hFis1 requires the endoplasmic reticulum gateway to induce apoptosis
Abstract
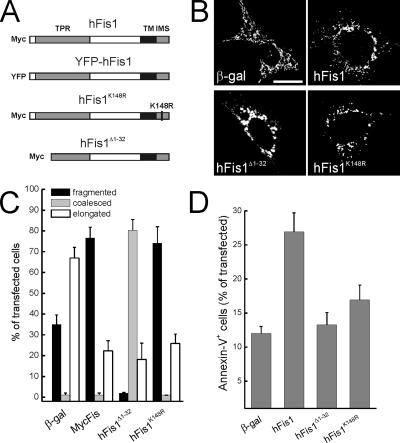
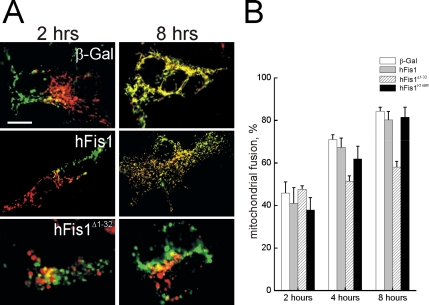
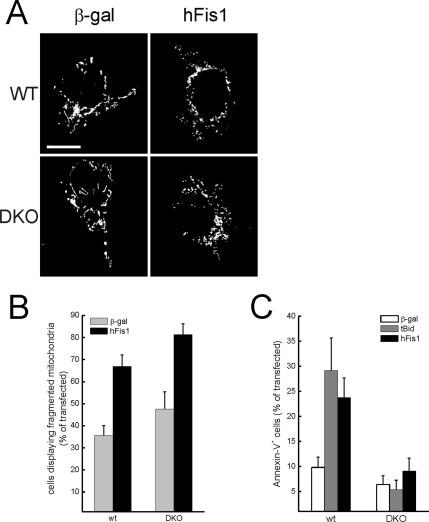
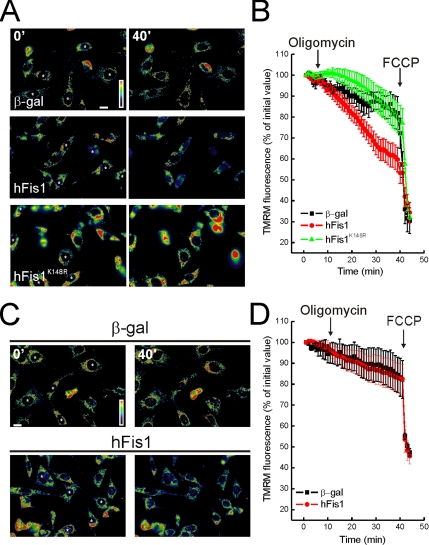
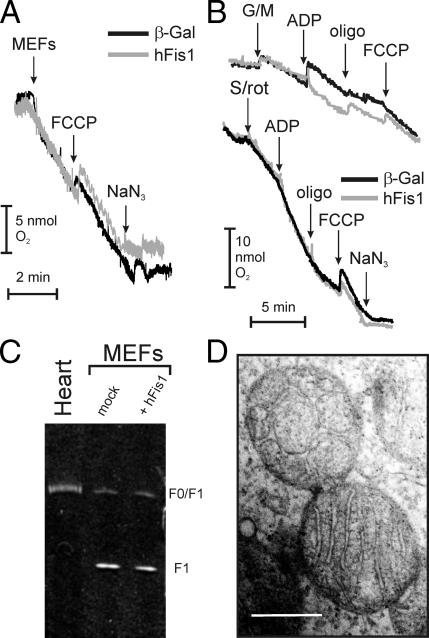
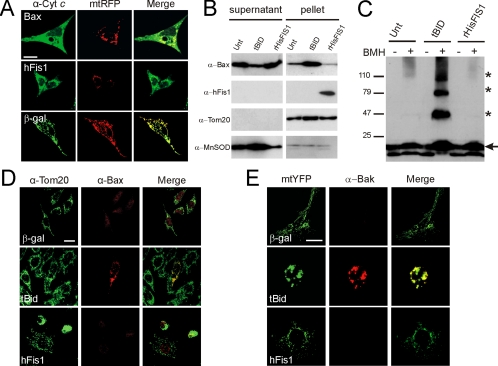
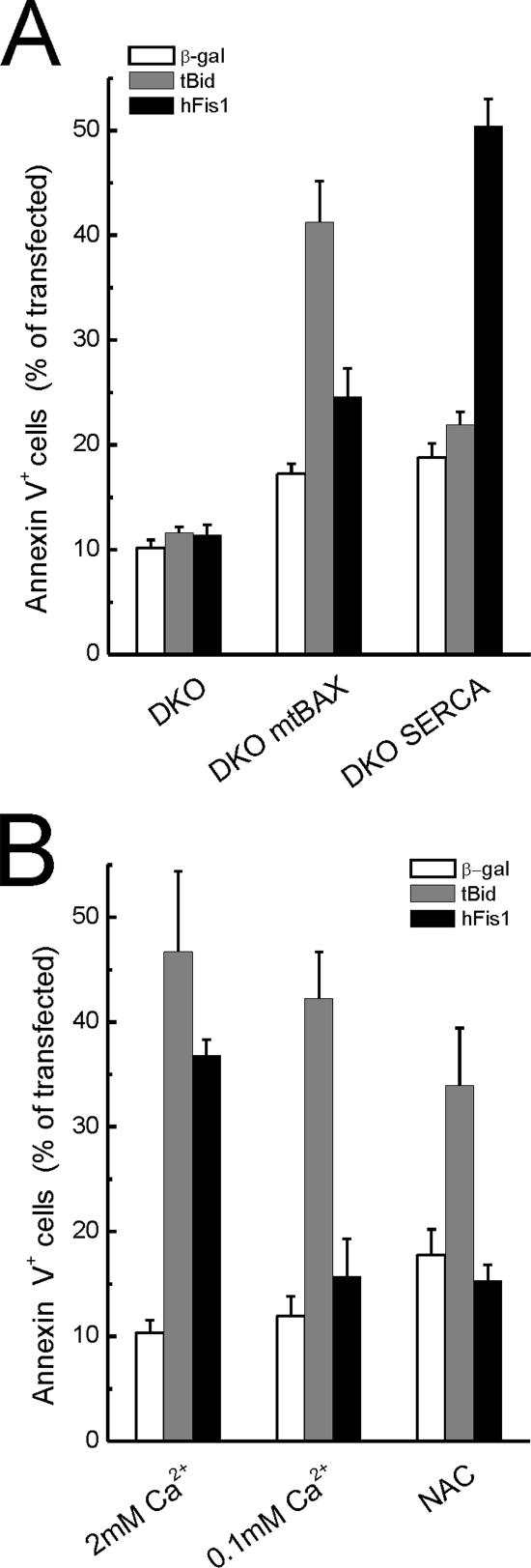
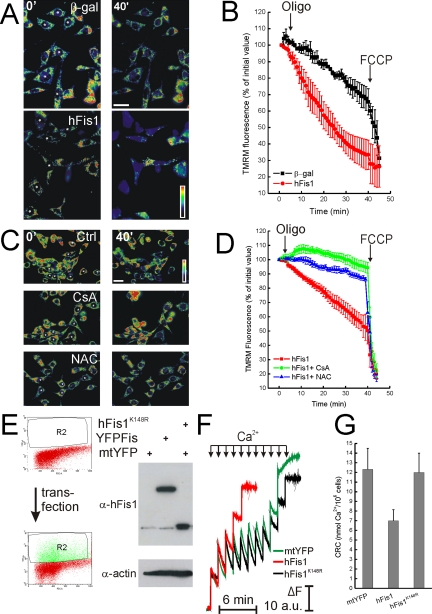
Mitochondrial fission ensures organelle inheritance during cell division and participates in apoptosis. The fission protein hFis1 triggers caspase-dependent cell death, by causing the release of cytochrome c from mitochondria. Here we show that mitochondrial fission induced by hFis1 is genetically distinct from apoptosis. In cells lacking the multidomain proapoptotic Bcl-2 family members Bax and Bak (DKO), hFis1 caused mitochondrial fragmentation but not organelle dysfunction and apoptosis. Similarly, a mutant in the intermembrane region of hFis1-induced fission but not cell death, further dissociating mitochondrial fragmentation from apoptosis induction. Selective correction of the endoplasmic reticulum (ER) defect of DKO cells restored killing by hFis1, indicating that death by hFis1 relies on the ER gateway of apoptosis. Consistently, hFis1 did not directly activate BAX and BAK, but induced Ca(2+)-dependent mitochondrial dysfunction. Thus, hFis1 is a bifunctional protein that independently regulates mitochondrial fragmentation and ER-mediated apoptosis.
Figures
References
-
- Arnoult D., Rismanchi N., Grodet A., Roberts R. G., Seeburg D. P., Estaquier J., Sheng M., Blackstone C. Bax/Bak-dependent release of DDP/TIMM8a promotes Drp1-mediated mitochondrial fission and mitoptosis during programmed cell death. Curr. Biol. 2005;15:2112–2118. - PubMed
-
- Bernardi P. Mitochondrial transport of cations: channels, exchangers and permeability transition. Physiol. Rev. 1999;79:1127–1155. - PubMed
-
- Bossy-Wetzel E., Barsoum M. J., Godzik A., Schwarzenbacher R., Lipton S. A. Mitochondrial fission in apoptosis, neurodegeneration and aging. Curr. Opin. Cell Biol. 2003;15:706–716. - PubMed
-
- Chen H., Chomyn A., Chan D. C. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J. Biol. Chem. 2005;280:26185–26192. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
