

A2A adenosine receptor protects tumors from antitumor T cells
- PMID: 16916931
- PMCID: PMC1559765
- DOI: 10.1073/pnas.0605251103
A2A adenosine receptor protects tumors from antitumor T cells
Abstract
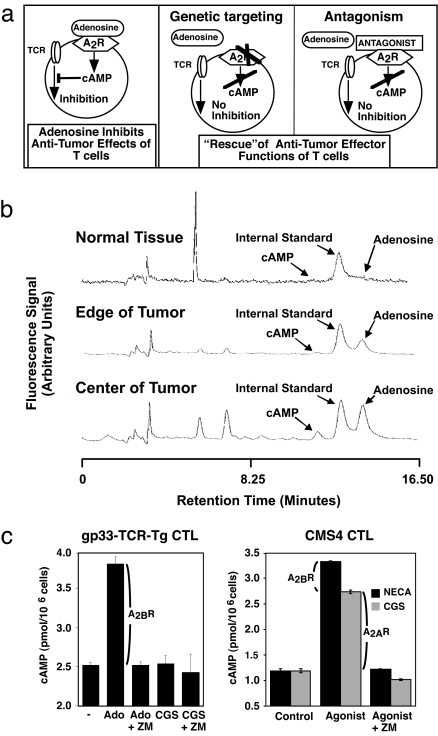
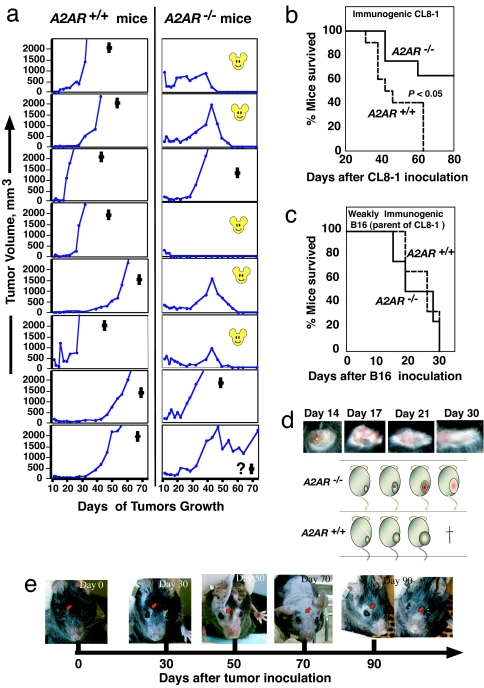
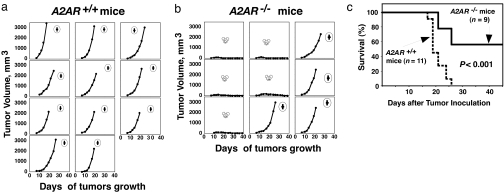
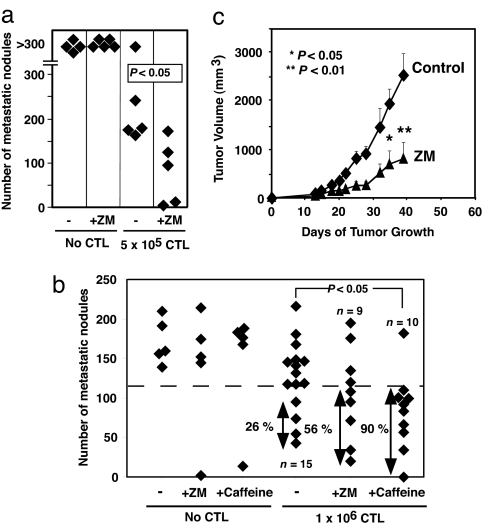
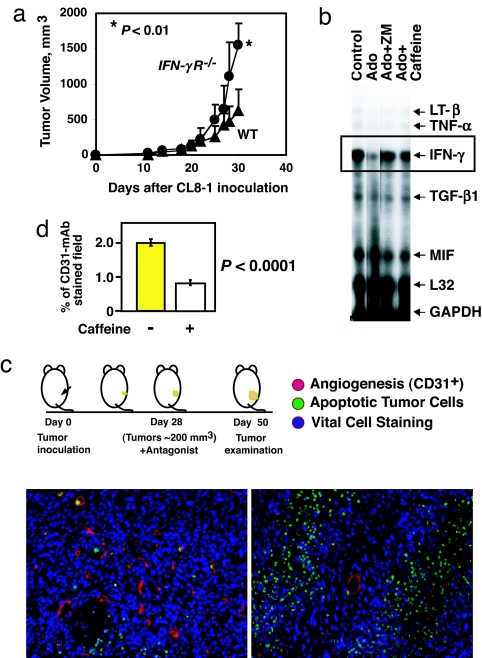
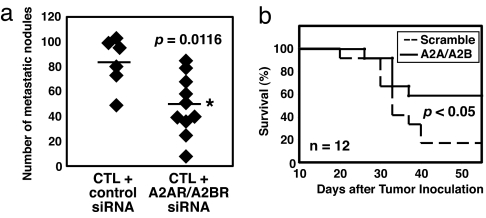
The A2A adenosine receptor (A2AR) has been shown to be a critical and nonredundant negative regulator of immune cells in protecting normal tissues from inflammatory damage. We hypothesized that A2AR also protects cancerous tissues by inhibiting incoming antitumor T lymphocytes. Here we confirm this hypothesis by showing that genetic deletion of A2AR in the host resulted in rejection of established immunogenic tumors in approximately 60% of A2AR-deficient mice with no rejection observed in control WT mice. The use of antagonists, including caffeine, or targeting the A2 receptors by siRNA pretreatment of T cells improved the inhibition of tumor growth, destruction of metastases, and prevention of neovascularization by antitumor T cells. The data suggest that effects of A2AR are T cell autonomous. The inhibition of antitumor T cells via their A2AR in the adenosine-rich tumor microenvironment may explain the paradoxical coexistence of tumors and antitumor immune cells in some cancer patients (the "Hellstrom paradox"). We propose to target the hypoxia-->adenosine-->A2AR pathway as a cancer immunotherapy strategy to prevent the inhibition of antitumor T cells in the tumor microenvironment. The same strategy may prevent the premature termination of immune response and improve the vaccine-induced development of antitumor and antiviral T cells. The observations of autoimmunity during melanoma rejection in A2AR-deficient mice suggest that A2AR in T cells is also important in preventing autoimmunity. Thus, although using the hypoxia-->adenosine-->A2AR pathway inhibitors may improve antitumor immunity, the recruitment of this pathway by selective drugs is expected to attenuate the autoimmune tissue damage.
Conflict of interest statement
Conflict of interest statement: No conflicts declared.
Figures
References
-
- Hellstrom I., Hellstrom K. E., Pierce G. E. Int. J. Cancer. 1968;3:467–482. - PubMed
-
- Hanson H. L., Donermeyer D. L., Ikeda H., White J. M., Shankaran V., Old L. J., Shiku H., Schreiber R. D., Allen P. M. Immunity. 2000;13:265–276. - PubMed
-
- Pardoll D., Allison J. Nat. Med. 2004:887–92. - PubMed
-
- Shankaran V., Ikeda H., Bruce A. T., White J. M., Swanson P. E., Old L. J., Schreiber R. D. Nature. 2001;410:1107–1111. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
