

Mass spectrometry-based proteomics and its application to studies of Porphyromonas gingivalis invasion and pathogenicity
- PMID: 16918489
- PMCID: PMC2666350
- DOI: 10.2174/187152606778249935
Mass spectrometry-based proteomics and its application to studies of Porphyromonas gingivalis invasion and pathogenicity
Abstract
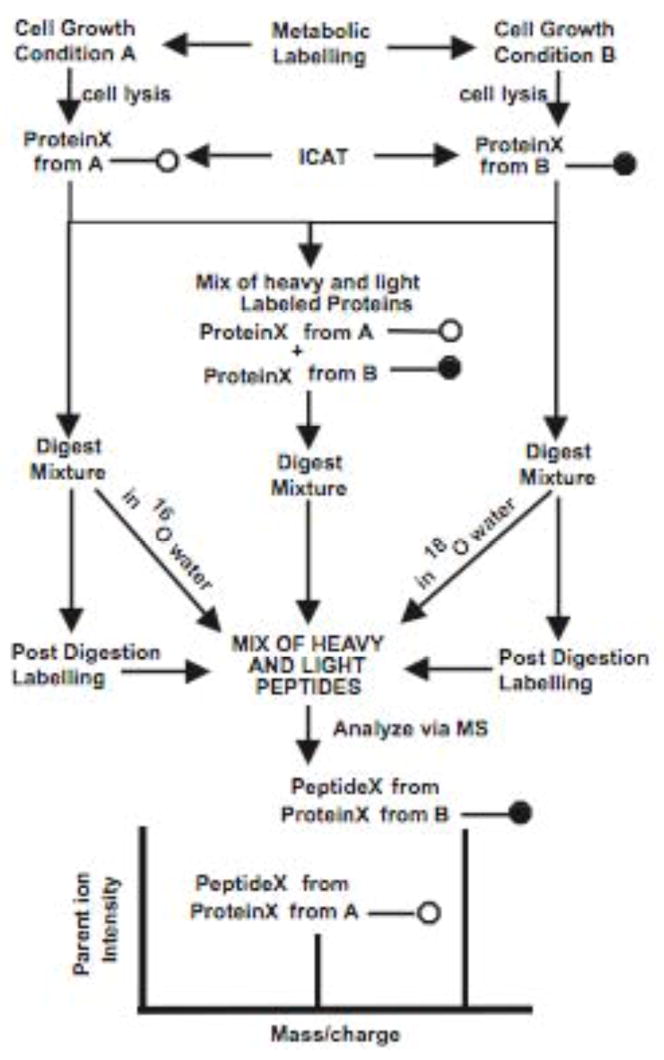
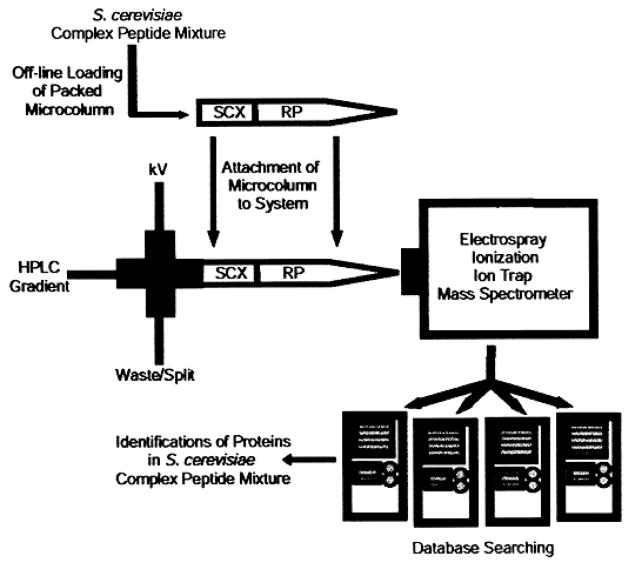
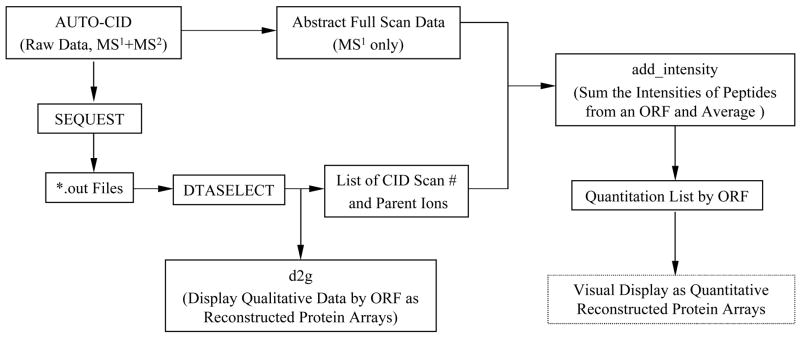
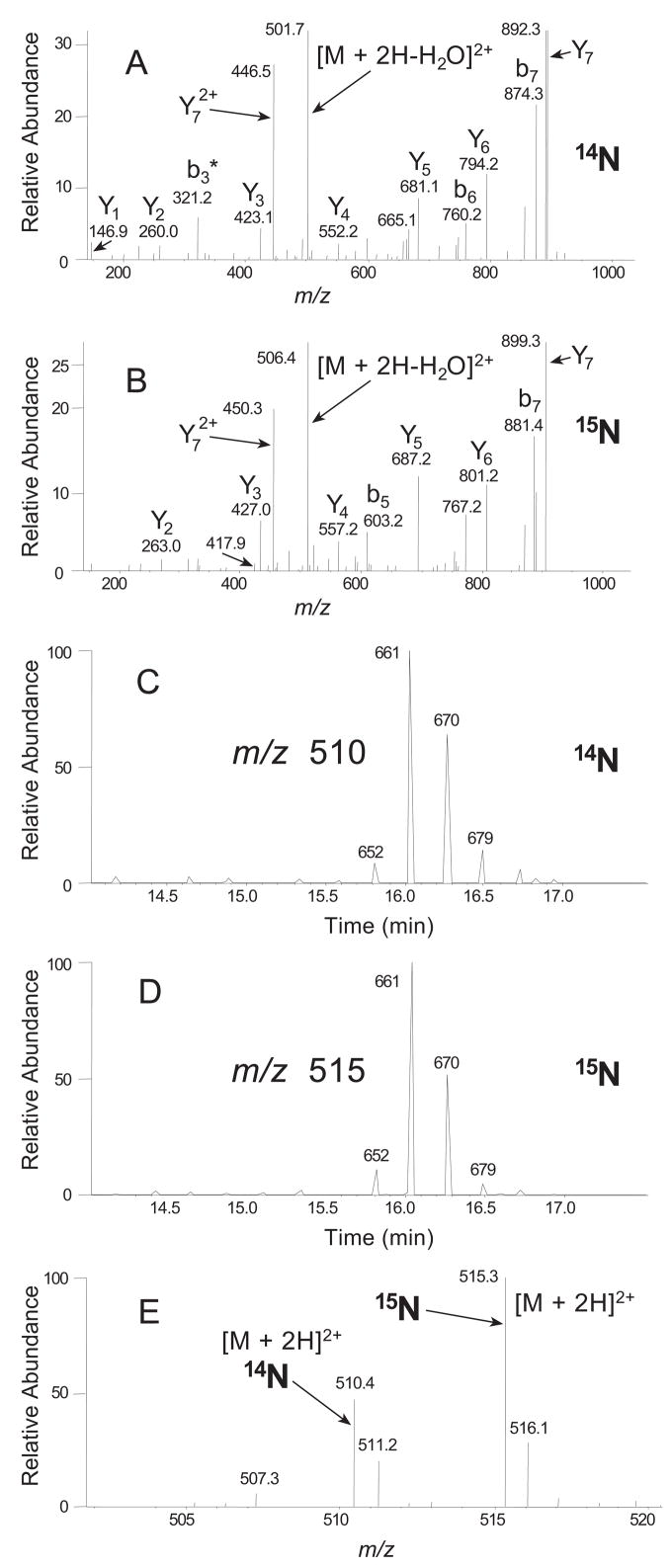
Porphyromonas gingivalis is a Gram-negative anaerobe that populates the subgingival crevice of the mouth. It is known to undergo a transition from its commensal status in healthy individuals to a highly invasive intracellular pathogen in human patients suffering from periodontal disease, where it is often the dominant species of pathogenic bacteria. The application of mass spectrometry-based proteomics to the study of P. gingivalis interactions with model host cell systems, invasion and pathogenicity is reviewed. These studies have evolved from qualitative identifications of small numbers of secreted proteins, using traditional gel-based methods, to quantitative whole cell proteomic studies using multiple dimension capillary HPLC coupled with linear ion trap mass spectrometry. It has become possible to generate a differential readout of protein expression change over the entire P. gingivalis proteome, in a manner analogous to whole genome mRNA arrays. Different strategies have been employed for generating protein level expression ratios from mass spectrometry data, including stable isotope metabolic labeling and most recently, spectral counting methods. A global view of changes in protein modification status remains elusive due to the limitations of existing computational tools for database searching and data mining. Such a view would be desirable for purposes of making global assessments of changes in gene regulation in response to host interactions during the course of adhesion, invasion and internalization. With a complete data matrix consisting of changes in transcription, protein abundance and protein modification during the course of invasion, the search for new protein drug targets would benefit from a more comprehensive understanding of these processes than what could be achieved prior to the advent of systems biology.
Figures

Similar articles
-
Pathway analysis for intracellular Porphyromonas gingivalis using a strain ATCC 33277 specific database.BMC Microbiol. 2009 Sep 1;9:185. doi: 10.1186/1471-2180-9-185. BMC Microbiol. 2009. PMID: 19723305 Free PMC article.
-
Label-free quantitative proteomic analysis of the oral bacteria Fusobacterium nucleatum and Porphyromonas gingivalis to identify protein features relevant in biofilm formation.Anaerobe. 2021 Dec;72:102449. doi: 10.1016/j.anaerobe.2021.102449. Epub 2021 Sep 17. Anaerobe. 2021. PMID: 34543761
-
Quantitative proteomics of intracellular Porphyromonas gingivalis.Proteomics. 2007 Dec;7(23):4323-37. doi: 10.1002/pmic.200700543. Proteomics. 2007. PMID: 17979175 Free PMC article.
-
Life below the gum line: pathogenic mechanisms of Porphyromonas gingivalis.Microbiol Mol Biol Rev. 1998 Dec;62(4):1244-63. doi: 10.1128/MMBR.62.4.1244-1263.1998. Microbiol Mol Biol Rev. 1998. PMID: 9841671 Free PMC article. Review.
-
Contribution of -Omics Technologies in the Study of Porphyromonas gingivalis during Periodontitis Pathogenesis: A Minireview.Int J Mol Sci. 2022 Dec 30;24(1):620. doi: 10.3390/ijms24010620. Int J Mol Sci. 2022. PMID: 36614064 Free PMC article. Review.
Cited by
-
What are We Learning and What Can We Learn from the Human Oral Microbiome Project?Curr Oral Health Rep. 2016 Mar;3(1):56-63. doi: 10.1007/s40496-016-0080-4. Epub 2016 Jan 23. Curr Oral Health Rep. 2016. PMID: 27152251 Free PMC article.
-
Tools for interpreting large-scale protein profiling in microbiology.J Dent Res. 2008 Nov;87(11):1004-15. doi: 10.1177/154405910808701113. J Dent Res. 2008. PMID: 18946006 Free PMC article. Review.
-
Cellular and bacterial profiles associated with oral epithelium-microbiota interactions.Periodontol 2000. 2010 Feb;52(1):207-17. doi: 10.1111/j.1600-0757.2009.00322.x. Periodontol 2000. 2010. PMID: 20017802 Free PMC article. Review. No abstract available.
-
Comparison of spectral counting and metabolic stable isotope labeling for use with quantitative microbial proteomics.Analyst. 2006 Dec;131(12):1335-41. doi: 10.1039/b610957h. Epub 2006 Oct 11. Analyst. 2006. PMID: 17124542 Free PMC article.
-
Heterogeneous Porphyromonas gingivalis LPS modulates immuno-inflammatory response, antioxidant defense and cytoskeletal dynamics in human gingival fibroblasts.Sci Rep. 2016 Aug 19;6:29829. doi: 10.1038/srep29829. Sci Rep. 2016. PMID: 27538450 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
