

GAA repeat expansion mutation mouse models of Friedreich ataxia exhibit oxidative stress leading to progressive neuronal and cardiac pathology
- PMID: 16919418
- PMCID: PMC2842930
- DOI: 10.1016/j.ygeno.2006.06.015
GAA repeat expansion mutation mouse models of Friedreich ataxia exhibit oxidative stress leading to progressive neuronal and cardiac pathology
Abstract
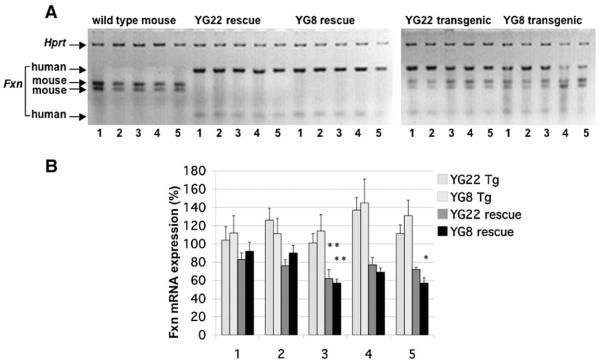
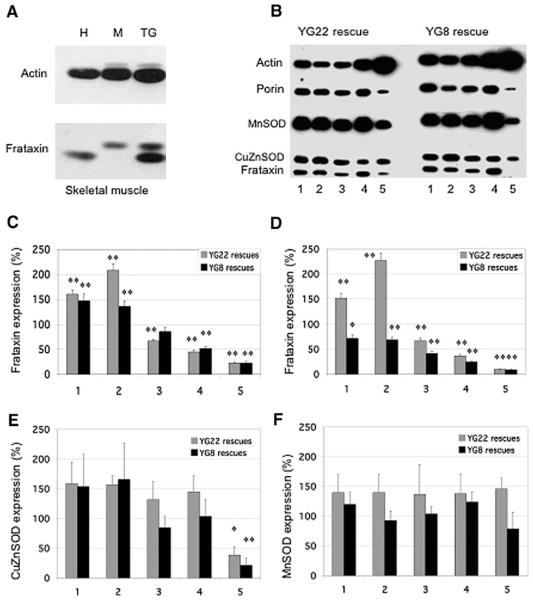
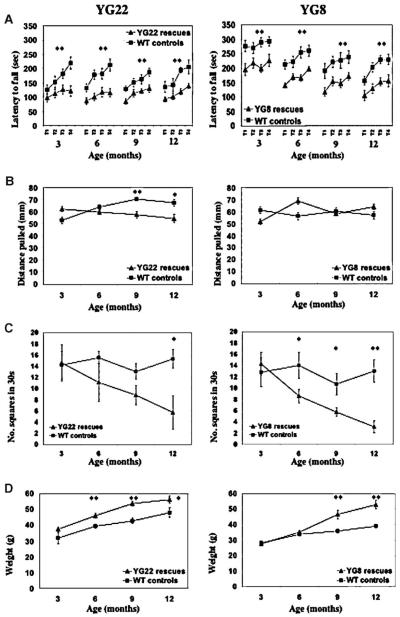
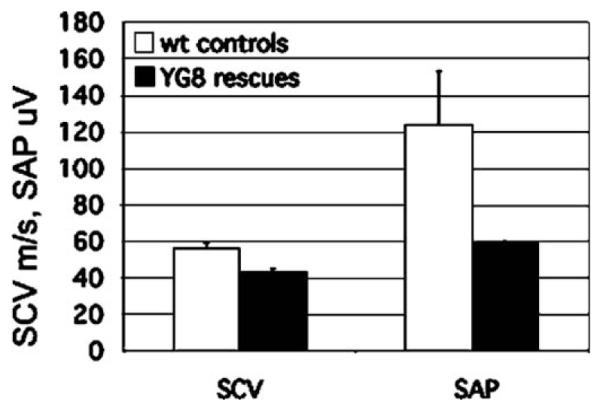
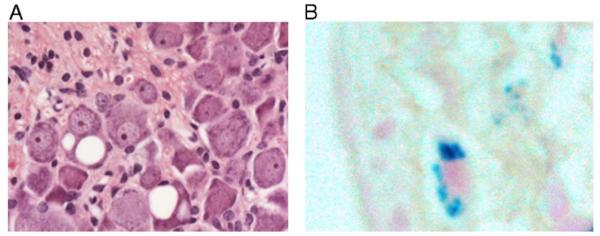
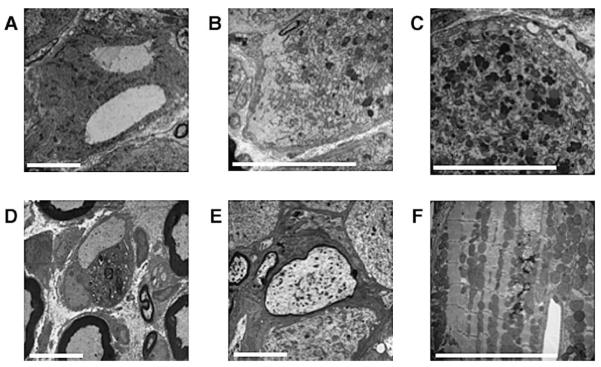
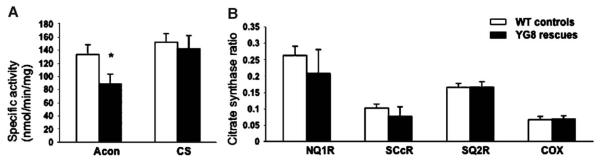
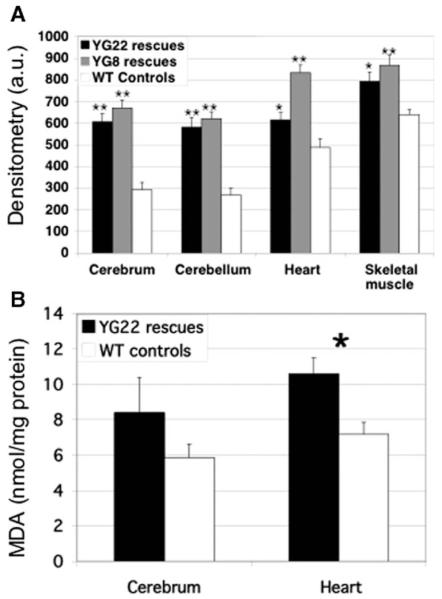
Friedreich ataxia (FRDA) is a neurodegenerative disorder caused by an unstable GAA repeat expansion mutation within intron 1 of the FXN gene. However, the origins of the GAA repeat expansion, its unstable dynamics within different cells and tissues, and its effects on frataxin expression are not yet completely understood. Therefore, we have chosen to generate representative FRDA mouse models by using the human FXN GAA repeat expansion itself as the genetically modified mutation. We have previously reported the establishment of two lines of human FXN YAC transgenic mice that contain unstable GAA repeat expansions within the appropriate genomic context. We now describe the generation of FRDA mouse models by crossbreeding of both lines of human FXN YAC transgenic mice with heterozygous Fxn knockout mice. The resultant FRDA mice that express only human-derived frataxin show comparatively reduced levels of frataxin mRNA and protein expression, decreased aconitase activity, and oxidative stress, leading to progressive neurodegenerative and cardiac pathological phenotypes. Coordination deficits are present, as measured by accelerating rotarod analysis, together with a progressive decrease in locomotor activity and increase in weight. Large vacuoles are detected within neurons of the dorsal root ganglia (DRG), predominantly within the lumbar regions in 6-month-old mice, but spreading to the cervical regions after 1 year of age. Secondary demyelination of large axons is also detected within the lumbar roots of older mice. Lipofuscin deposition is increased in both DRG neurons and cardiomyocytes, and iron deposition is detected in cardiomyocytes after 1 year of age. These mice represent the first GAA repeat expansion-based FRDA mouse models that exhibit progressive FRDA-like pathology and thus will be of use in testing potential therapeutic strategies, particularly GAA repeat-based strategies.
Figures
References
-
- Campuzano V, Montermini L, Molto MD, Pianese L, Cossee M, Cavalcanti F, Monros E, Rodius F, Duclos F, Monticelli A, Zara F, Canizares J, Koutnikova H, Bidichandani SI, Gellera C, Brice A, Trouillas P, De Michele G, Filla A, De Frutos R, Palau F, Patel PI, Di Donato S, Mandel JL, Cocozza S, Koenig M, Pandolfo M. Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science. 1996;271:1423–1427. - PubMed
-
- Pandolfo M. The molecular basis of Friedreich ataxia. Adv. Exp. Med. Biol. 2002;516:99–118. - PubMed
-
- Campuzano V, Montermini L, Lutz Y, Cova L, Hindelang C, Jiralerspong S, Trottier Y, Kish SJ, Faucheux B, Trouillas P, Authier FJ, Durr A, Mandel JL, Vescovi A, Pandolfo M, Koenig M. Frataxin is reduced in Friedreich ataxia patients and is associated with mitochondrial membranes. Hum. Mol. Genet. 1997;6:1771–1780. - PubMed
-
- Gonzalez-Cabo P, Vazquez-Manrique RP, Garcia-Gimeno MA, Sanz P, Palau F. Frataxin interacts functionally with mitochondrial electron transport chain proteins. Hum. Mol. Genet. 2005;14:2091–2098. - PubMed
-
- Bulteau AL, O’Neill HA, Kennedy MC, Ikeda-Saito M, Isaya G, Szweda LI. Frataxin acts as an iron chaperone protein to modulate mitochondrial aconitase activity. Science. 2004;305:242–245. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
