EGASP: the human ENCODE Genome Annotation Assessment Project
- PMID: 16925836
- PMCID: PMC1810551
- DOI: 10.1186/gb-2006-7-s1-s2
EGASP: the human ENCODE Genome Annotation Assessment Project
Abstract
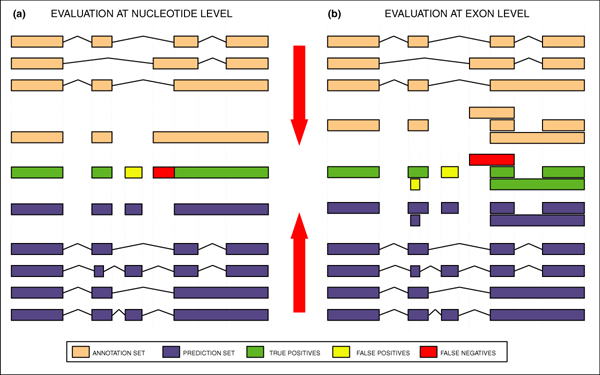
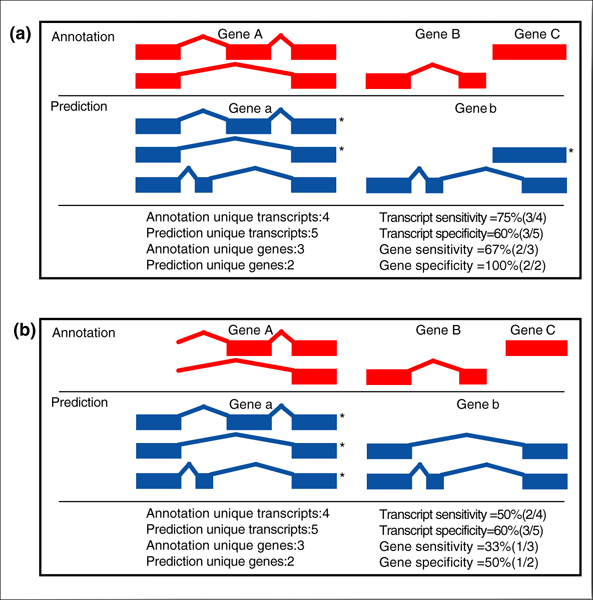
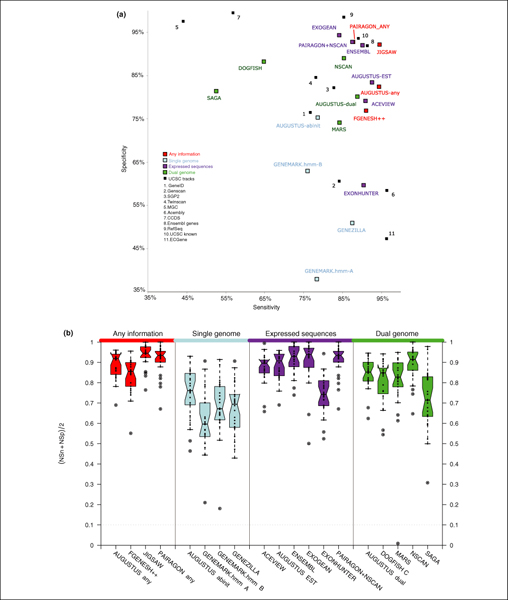
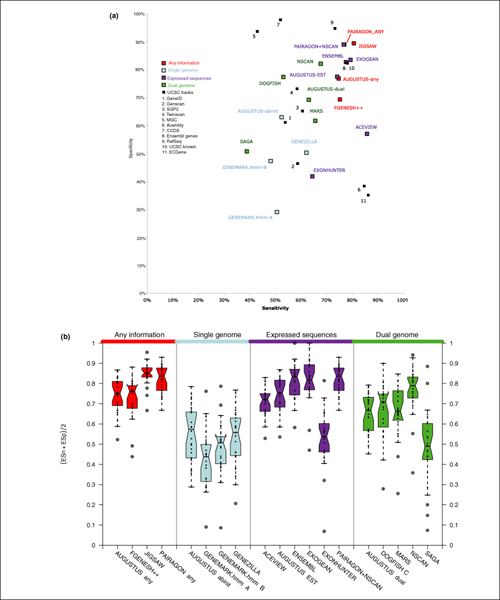
Background: We present the results of EGASP, a community experiment to assess the state-of-the-art in genome annotation within the ENCODE regions, which span 1% of the human genome sequence. The experiment had two major goals: the assessment of the accuracy of computational methods to predict protein coding genes; and the overall assessment of the completeness of the current human genome annotations as represented in the ENCODE regions. For the computational prediction assessment, eighteen groups contributed gene predictions. We evaluated these submissions against each other based on a 'reference set' of annotations generated as part of the GENCODE project. These annotations were not available to the prediction groups prior to the submission deadline, so that their predictions were blind and an external advisory committee could perform a fair assessment.
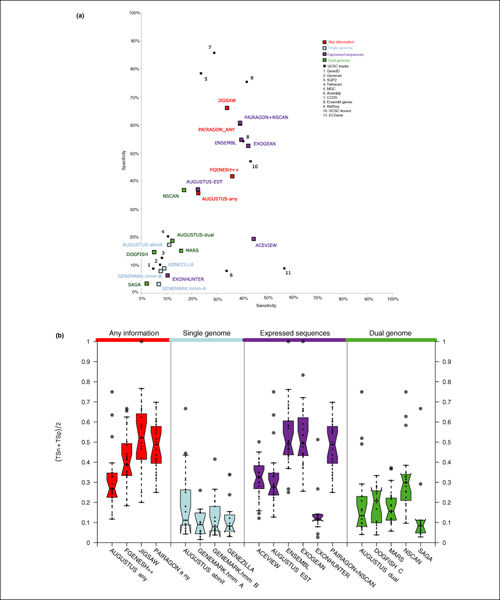
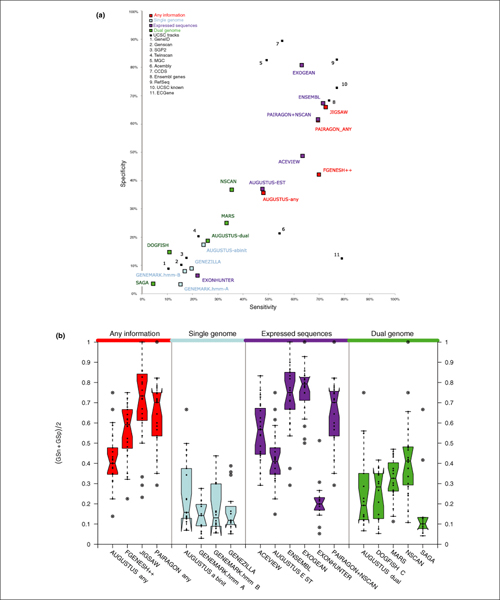
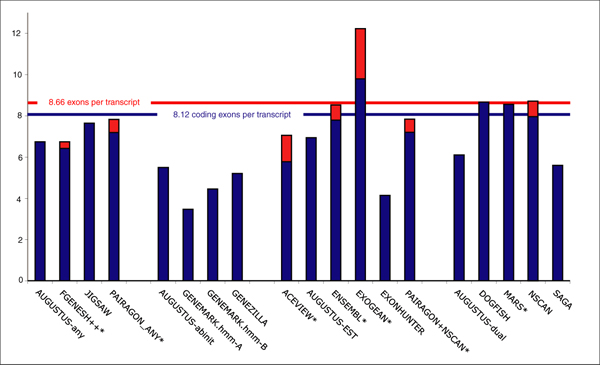
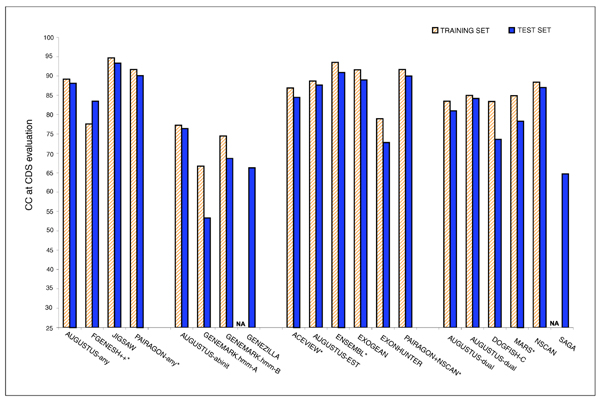
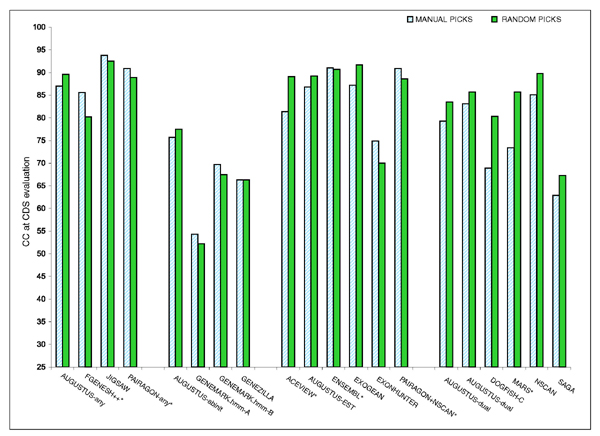
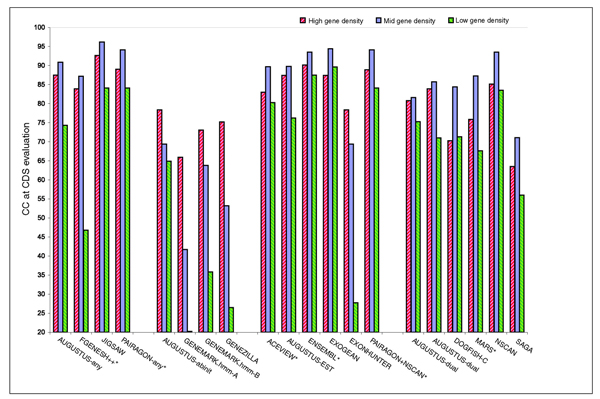
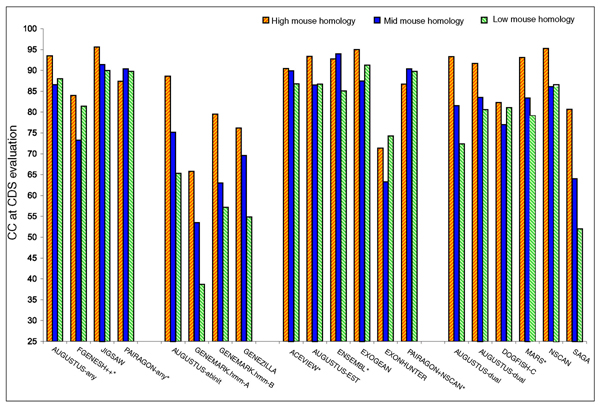
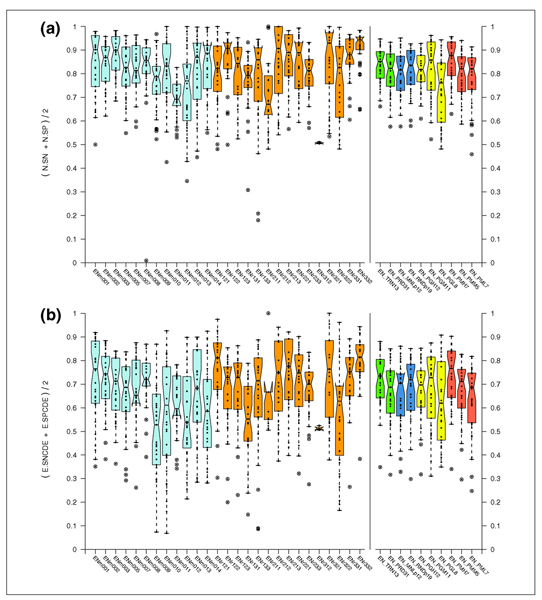
Results: The best methods had at least one gene transcript correctly predicted for close to 70% of the annotated genes. Nevertheless, the multiple transcript accuracy, taking into account alternative splicing, reached only approximately 40% to 50% accuracy. At the coding nucleotide level, the best programs reached an accuracy of 90% in both sensitivity and specificity. Programs relying on mRNA and protein sequences were the most accurate in reproducing the manually curated annotations. Experimental validation shows that only a very small percentage (3.2%) of the selected 221 computationally predicted exons outside of the existing annotation could be verified.
Conclusion: This is the first such experiment in human DNA, and we have followed the standards established in a similar experiment, GASP1, in Drosophila melanogaster. We believe the results presented here contribute to the value of ongoing large-scale annotation projects and should guide further experimental methods when being scaled up to the entire human genome sequence.
Figures