

Performance assessment of promoter predictions on ENCODE regions in the EGASP experiment
- PMID: 16925837
- PMCID: PMC1810552
- DOI: 10.1186/gb-2006-7-s1-s3
Performance assessment of promoter predictions on ENCODE regions in the EGASP experiment
Abstract
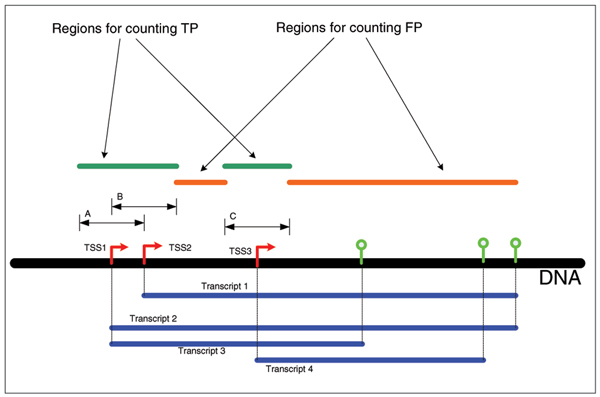
Background: This study analyzes the predictions of a number of promoter predictors on the ENCODE regions of the human genome as part of the ENCODE Genome Annotation Assessment Project (EGASP). The systems analyzed operate on various principles and we assessed the effectiveness of different conceptual strategies used to correlate produced promoter predictions with the manually annotated 5' gene ends.
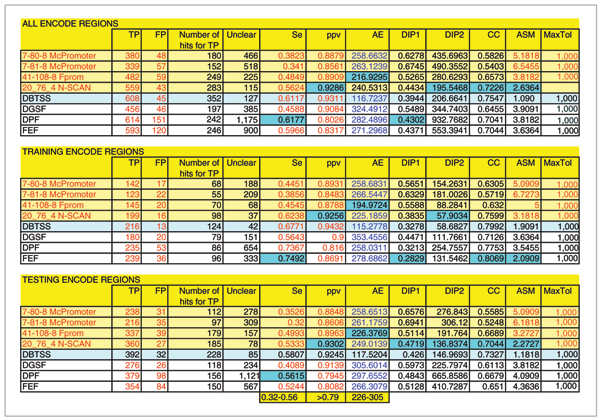
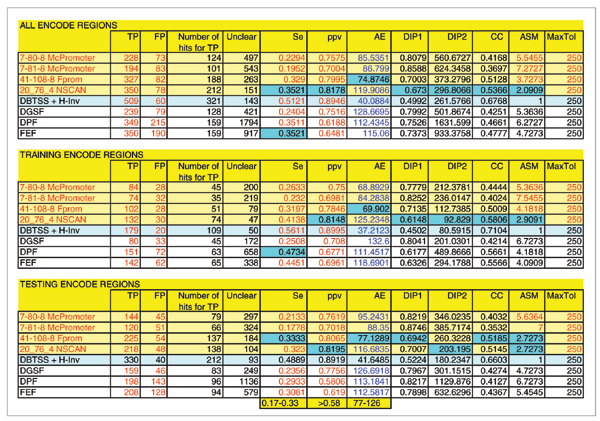
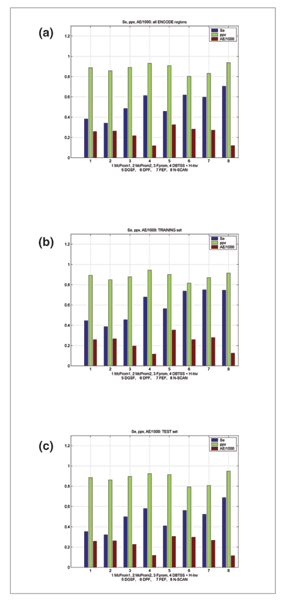
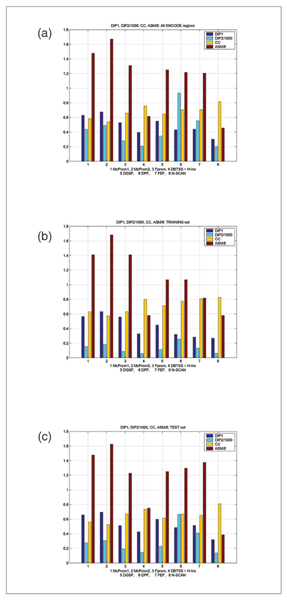
Results: The predictions were assessed relative to the manual HAVANA annotation of the 5' gene ends. These 5' gene ends were used as the estimated reference transcription start sites. With the maximum allowed distance for predictions of 1,000 nucleotides from the reference transcription start sites, the sensitivity of predictors was in the range 32% to 56%, while the positive predictive value was in the range 79% to 93%. The average distance mismatch of predictions from the reference transcription start sites was in the range 259 to 305 nucleotides. At the same time, using transcription start site estimates from DBTSS and H-Invitational databases as promoter predictions, we obtained a sensitivity of 58%, a positive predictive value of 92%, and an average distance from the annotated transcription start sites of 117 nucleotides. In this experiment, the best performing promoter predictors were those that combined promoter prediction with gene prediction. The main reason for this is the reduced promoter search space that resulted in smaller numbers of false positive predictions.
Conclusion: The main finding, now supported by comprehensive data, is that the accuracy of human promoter predictors for high-throughput annotation purposes can be significantly improved if promoter prediction is combined with gene prediction. Based on the lessons learned in this experiment, we propose a framework for the preparation of the next similar promoter prediction assessment.
Figures
References
-
- Weinzierl ROJ. Mechanisms of Gene Expression: Structure, Function, and Evolution of the Basal Transcriptional Machinery. London: Imperial College Press; 1999.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
