

Pairagon+N-SCAN_EST: a model-based gene annotation pipeline
- PMID: 16925839
- PMCID: PMC1810554
- DOI: 10.1186/gb-2006-7-s1-s5
Pairagon+N-SCAN_EST: a model-based gene annotation pipeline
Abstract
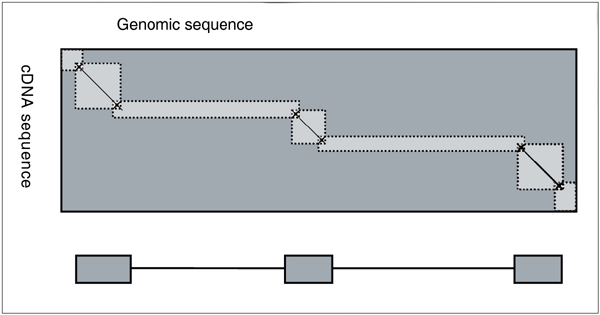
Background: This paper describes Pairagon+N-SCAN_EST, a gene annotation pipeline that uses only native alignments. For each expressed sequence it chooses the best genomic alignment. Systems like ENSEMBL and ExoGean rely on trans alignments, in which expressed sequences are aligned to the genomic loci of putative homologs. Trans alignments contain a high proportion of mismatches, gaps, and/or apparently unspliceable introns, compared to alignments of cDNA sequences to their native loci. The Pairagon+N-SCAN_EST pipeline's first stage is Pairagon, a cDNA-to-genome alignment program based on a PairHMM probability model. This model relies on prior knowledge, such as the fact that introns must begin with GT, GC, or AT and end with AG or AC. It produces very precise alignments of high quality cDNA sequences. In the genomic regions between Pairagon's cDNA alignments, the pipeline combines EST alignments with de novo gene prediction by using N-SCAN_EST. N-SCAN_EST is based on a generalized HMM probability model augmented with a phylogenetic conservation model and EST alignments. It can predict complete transcripts by extending or merging EST alignments, but it can also predict genes in regions without EST alignments. Because they are based on probability models, both Pairagon and N-SCAN_EST can be trained automatically for new genomes and data sets.
Results: On the ENCODE regions of the human genome, Pairagon+N-SCAN_EST was as accurate as any other system tested in the EGASP assessment, including ENSEMBL and ExoGean.
Conclusion: With sufficient mRNA/EST evidence, genome annotation without trans alignments can compete successfully with systems like ENSEMBL and ExoGean, which use trans alignments.
Figures






References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
