

Idiopathic pulmonary fibrosis : new concepts in pathogenesis and implications for drug therapy
- PMID: 16928146
- PMCID: PMC2231521
- DOI: 10.2165/00151829-200605050-00004
Idiopathic pulmonary fibrosis : new concepts in pathogenesis and implications for drug therapy
Abstract
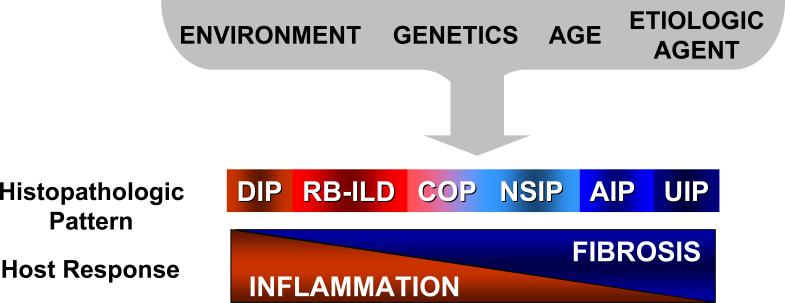
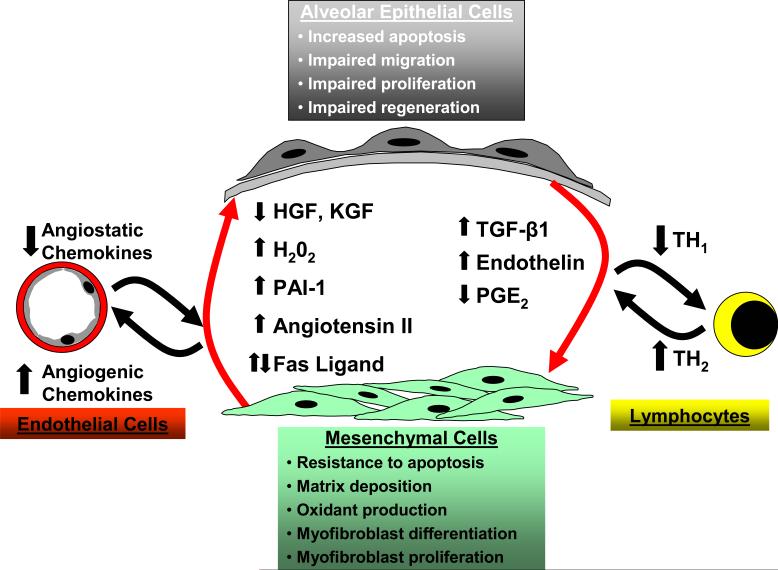
Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive, and usually fatal pulmonary disease for which there are no proven drug therapies. Anti-inflammatory and immunosuppressive agents have been largely ineffective. The precise relationship of IPF to other idiopathic interstitial pneumonias (IIPs) is not known, despite the observation that different histopathologic patterns of IIP may coexist in the same patient. We propose that these different histopathologic 'reaction' patterns may be determined by complex interactions between host and environmental factors that alter the local alveolar milieu. Recent paradigms in IPF pathogenesis have focused on dysregulated epithelial-mesenchymal interactions, an imbalance in T(H)1/T(H)2 cytokine profile and potential roles for aberrant angiogenesis. In this review, we discuss these evolving concepts in disease pathogenesis and emerging therapies designed to target pro-fibrogenic pathways in IPF.
Figures
References
-
- Schwartz DA, Helmers RA, Galvin JR, et al. Determinants of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1994;149(2 Pt 1):450–4. - PubMed
-
- King TE, Jr., Tooze JA, Schwarz MI, et al. Predicting survival in idiopathic pulmonary fibrosis: scoring system and survival model. Am J Respir Crit Care Med. 2001;164(7):1171–81. - PubMed
-
- American Thoracic Society Idiopathic pulmonary fibrosis: diagnosis and treatment. International consensus statement. American Thoracic Society (ATS), and the European Respiratory Society (ERS). Am J Respir Crit Care Med. 2000;161(2 Pt 1):646–64. - PubMed
-
- Coultas DB, Zumwalt RE, Black WC, et al. The epidemiology of interstitial lung diseases. Am J Respir Crit Care Med. 1994;150(4):967–72. - PubMed
-
- Lama VN, Flaherty KR, Toews GB, et al. Prognostic value of desaturation during a 6-minute walk test in idiopathic interstitial pneumonia. Am J Respir Crit Care Med. 2003;168(9):1084–90. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources