

The Chk1/Cdc25A pathway as activators of the cell cycle in neuronal death induced by camptothecin
- PMID: 16928871
- PMCID: PMC6674376
- DOI: 10.1523/JNEUROSCI.2593-06.2006
The Chk1/Cdc25A pathway as activators of the cell cycle in neuronal death induced by camptothecin
Abstract
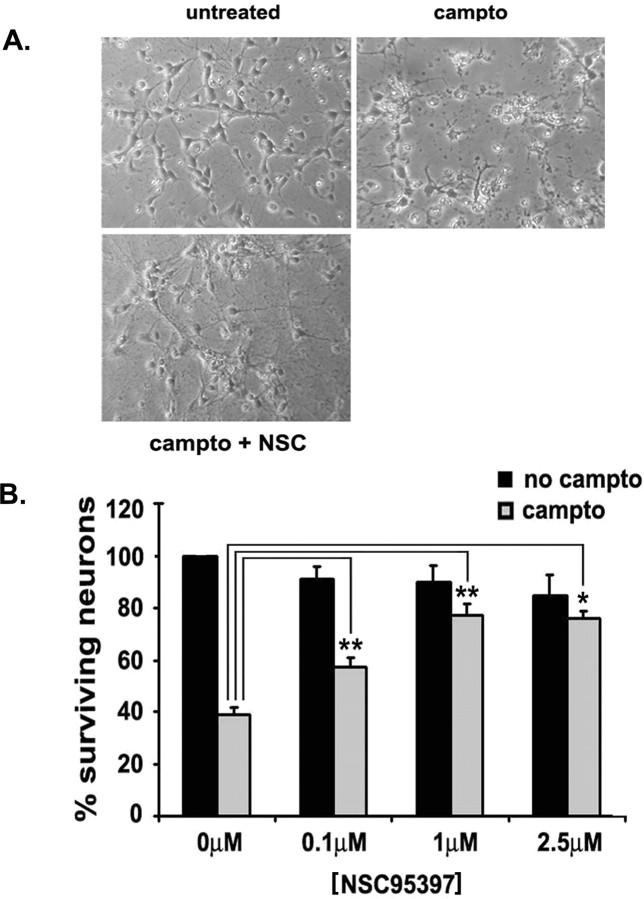
Cell cycle regulators appear to play a paradoxical role in neuronal death. We have shown previously that cyclin-dependent kinases (CDKs), along with their downstream effectors, Rb (retinoblastoma) and E2F/DP1 (E2 promoter binding factor/deleted in polyposis 1), regulate neuronal death evoked by the DNA damaging agent camptothecin. However, the mechanism by which CDKs are activated in this model is unclear. The cell division cycle 25A (Cdc25A) phosphatase is a critical regulator of cell cycle CDKs in proliferating cells. In cortical neurons, we presently show that expression of Cdc25A promotes death even in the absence of DNA damage. Importantly, Cdc25A activity is rapidly increased during DNA damage treatment. Inhibition of Cdc25A blocks death and reduces cyclin D1-associated kinase activity and Rb phosphorylation. This indicates that endogenous Cdc25A activity is important for regulation of cell cycle-mediated neuronal death. We also examined how Cdc25A activity is regulated after DNA damage. Cultured embryonic cortical neurons have a significant basal activity of checkpoint kinase 1 (Chk1), a kinase that regulates cell cycle arrest. During camptothecin treatment of neurons, this activity is rapidly downregulated with a concomitant increase in Cdc25A activity. Importantly, expression of wild-type Chk1, but not kinase-dead Chk1, inhibits the camptothecin-induced increase in Cdc25A activity. In addition, Chk1 expression also promotes survival in the presence of the DNA-damaging agent. Together, our data suggest that a Chk1/Cdc25A activity participates in activation of a cell cycle pathway-mediated death signal in neurons. These data also define how a proliferative signal may be abnormally activated in a postmitotic environment.
Figures







References
-
- Alam ZI, Jenner A, Daniel SE, Lees AJ, Cairns N, Marsden CD, Jenner P, Halliwell B. Oxidative DNA damage in the parkinsonian brain: an apparent selective increase in 8-hydroxyguanine levels in substantia nigra. J Neurochem. 1997;69:1196–1203. - PubMed
-
- Baratte B, Meijer L, Galaktionov K, Beach D. Screening for antimitotic compounds using the cdc25 tyrosine phosphatase, an activator of the mitosis-inducing p34cdc2/cyclin Bcdc13 protein kinase. Anticancer Res. 1992;12:873–880. - PubMed
-
- Becker EB, Bonni A. Cell cycle regulation of neuronal apoptosis in development and disease. Prog Neurobiol. 2004;72:1–25. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous