

PET-Tool: a software suite for comprehensive processing and managing of Paired-End diTag (PET) sequence data
- PMID: 16934139
- PMCID: PMC1564156
- DOI: 10.1186/1471-2105-7-390
PET-Tool: a software suite for comprehensive processing and managing of Paired-End diTag (PET) sequence data
Abstract
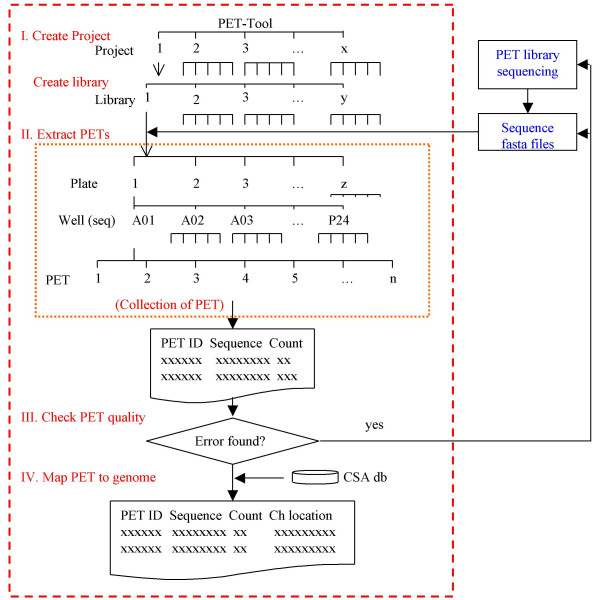
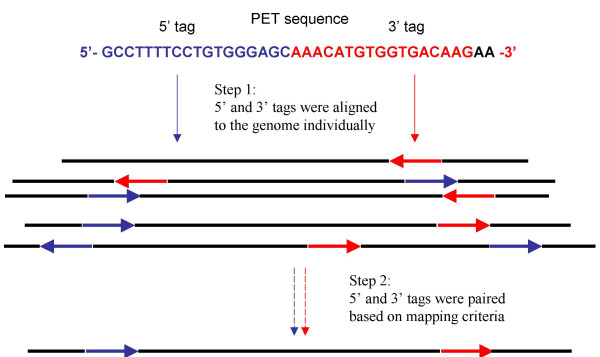
Background: We recently developed the Paired End diTag (PET) strategy for efficient characterization of mammalian transcriptomes and genomes. The paired end nature of short PET sequences derived from long DNA fragments raised a new set of bioinformatics challenges, including how to extract PETs from raw sequence reads, and correctly yet efficiently map PETs to reference genome sequences. To accommodate and streamline data analysis of the large volume PET sequences generated from each PET experiment, an automated PET data process pipeline is desirable.
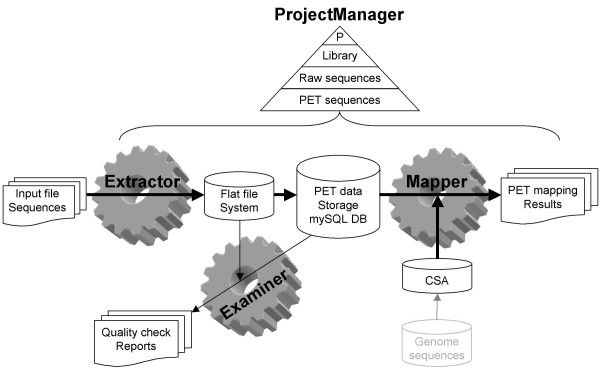
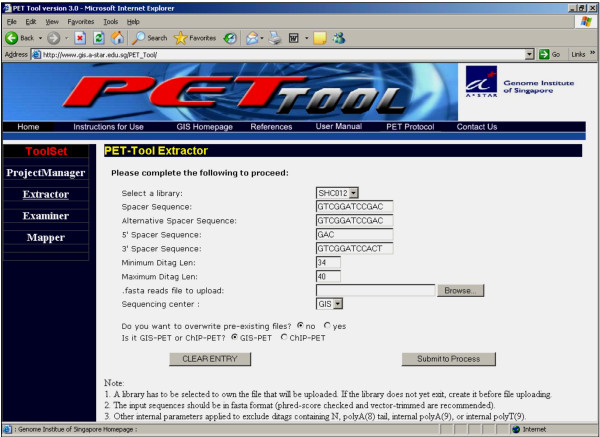
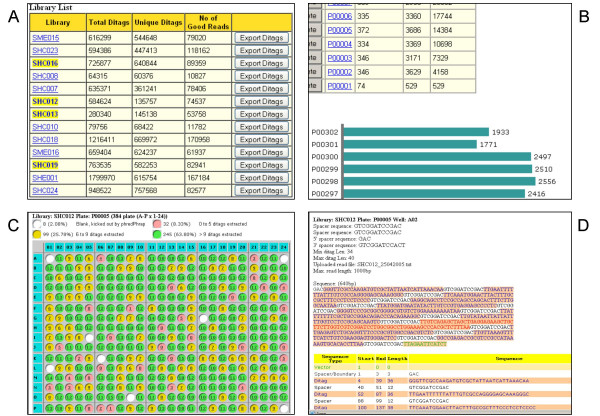
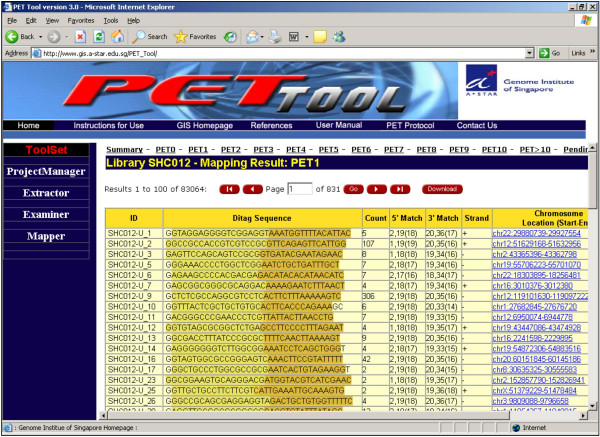
Results: We designed an integrated computation program package, PET-Tool, to automatically process PET sequences and map them to the genome sequences. The Tool was implemented as a web-based application composed of four modules: the Extractor module for PET extraction; the Examiner module for analytic evaluation of PET sequence quality; the Mapper module for locating PET sequences in the genome sequences; and the Project Manager module for data organization. The performance of PET-Tool was evaluated through the analyses of 2.7 million PET sequences. It was demonstrated that PET-Tool is accurate and efficient in extracting PET sequences and removing artifacts from large volume dataset. Using optimized mapping criteria, over 70% of quality PET sequences were mapped specifically to the genome sequences. With a 2.4 GHz LINUX machine, it takes approximately six hours to process one million PETs from extraction to mapping.
Conclusion: The speed, accuracy, and comprehensiveness have proved that PET-Tool is an important and useful component in PET experiments, and can be extended to accommodate other related analyses of paired-end sequences. The Tool also provides user-friendly functions for data quality check and system for multi-layer data management.
Figures
References
-
- Velculescu VE, Zhang L, Vogelstein B, Kinzler KW. Serial analysis of gene expression. Science. 1995;270:484–487. - PubMed
-
- Shiraki T, Kondo S, Katayama S, Waki K, Kasukawa T, Kawaji H, Kodzius R, Watahiki A, Nakamura M, Arakawa T, Fukuda S, Sasaki D, Podhajska A, Harbers M, Kawai J, Carninci P, Hayashizaki Y. Cap analysis gene expression for high-throughput analysis of transcriptional starting point and identification of promoter usage. PNAS USA. 2003;100:15776–15781. doi: 10.1073/pnas.2136655100. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
