

Selective leukemic-cell killing by a novel functional class of thalidomide analogs
- PMID: 16940421
- PMCID: PMC1895447
- DOI: 10.1182/blood-2006-04-017046
Selective leukemic-cell killing by a novel functional class of thalidomide analogs
Abstract
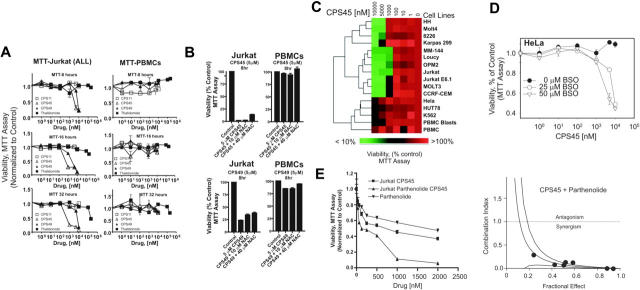
Using a novel cell-based assay to profile transcriptional pathway targeting, we have identified a new functional class of thalidomide analogs with distinct and selective antileukemic activity. These agents activate nuclear factor of activated T cells (NFAT) transcriptional pathways while simultaneously repressing nuclear factor-kappaB (NF-kappaB) via a rapid intracellular amplification of reactive oxygen species (ROS). The elevated ROS is associated with increased intracellular free calcium, rapid dissipation of the mitochondrial membrane potential, disrupted mitochondrial structure, and caspase-independent cell death. This cytotoxicity is highly selective for transformed lymphoid cells, is reversed by free radical scavengers, synergizes with the antileukemic activity of other redox-directed compounds, and preferentially targets cells in the S phase of the cell cycle. Live-cell imaging reveals a rapid drug-induced burst of ROS originating in the endoplasmic reticulum and associated mitochondria just prior to spreading throughout the cell. As members of a novel functional class of "redoxreactive" thalidomides, these compounds provide a new tool through which selective cellular properties of redox status and intracellular bioactivation can be leveraged by rational combinatorial therapeutic strategies and appropriate drug design to exploit cell-specific vulnerabilities for maximum drug efficacy.
Figures






Similar articles
-
NF-kappaB inhibitors from Brucea javanica exhibiting intracellular effects on reactive oxygen species.Anticancer Res. 2010 Sep;30(9):3295-300. Anticancer Res. 2010. PMID: 20944100 Free PMC article.
-
Combinatorial antileukemic disruption of oxidative homeostasis and mitochondrial stability by the redox reactive thalidomide 2-(2,4-difluoro-phenyl)-4,5,6,7-tetrafluoro-1H-isoindole-1,3(2H)-dione (CPS49) and flavopiridol.Mol Pharmacol. 2008 Sep;74(3):872-83. doi: 10.1124/mol.107.040808. Epub 2008 Jun 12. Mol Pharmacol. 2008. PMID: 18556456 Free PMC article.
-
Exposure to the anti-TNF-alpha drug thalidomide induces apoptotic cell death in human T leukemic cells.Cell Mol Biol (Noisy-le-grand). 2003 Nov;49(7):1117-24. Cell Mol Biol (Noisy-le-grand). 2003. PMID: 14682394
-
Targeting Oncogenic Nuclear Factor Kappa B Signaling with Redox-Active Agents for Cancer Treatment.Antioxid Redox Signal. 2019 Mar 10;30(8):1096-1123. doi: 10.1089/ars.2017.7387. Epub 2018 Feb 6. Antioxid Redox Signal. 2019. PMID: 29161883 Review.
-
Immunomodulatory drugs.Cancer Invest. 2005;23(7):625-34. doi: 10.1080/07357900500283101. Cancer Invest. 2005. PMID: 16305990 Review.
Cited by
-
Thalidomide analogues as anticancer drugs.Recent Pat Anticancer Drug Discov. 2007 Jun;2(2):167-74. doi: 10.2174/157489207780832478. Recent Pat Anticancer Drug Discov. 2007. PMID: 17975653 Free PMC article. Review.
-
N-benzoxazol-2-yl-N'-1-(isoquinolin-3-yl-ethylidene)-hydrazine, a novel compound with antitumor activity, induces radicals and dissipation of mitochondrial membrane potential.Invest New Drugs. 2009 Jun;27(3):189-202. doi: 10.1007/s10637-008-9156-x. Epub 2008 Jul 9. Invest New Drugs. 2009. PMID: 18612590
-
Mechanism of immunomodulatory drugs' action in the treatment of multiple myeloma.Acta Biochim Biophys Sin (Shanghai). 2014 Mar;46(3):240-53. doi: 10.1093/abbs/gmt142. Epub 2013 Dec 29. Acta Biochim Biophys Sin (Shanghai). 2014. PMID: 24374776 Free PMC article. Review.
-
NF-kappaB inhibitors from Brucea javanica exhibiting intracellular effects on reactive oxygen species.Anticancer Res. 2010 Sep;30(9):3295-300. Anticancer Res. 2010. PMID: 20944100 Free PMC article.
-
Polyunsaturated fatty acids synergize with lipid droplet binding thalidomide analogs to induce oxidative stress in cancer cells.Lipids Health Dis. 2010 Jun 2;9:56. doi: 10.1186/1476-511X-9-56. Lipids Health Dis. 2010. PMID: 20525221 Free PMC article.
References
-
- Franks ME, Macpherson GR, Figg WD. Thalidomide. Lancet. 2004;363: 1802-1811. - PubMed
-
- Bartlett JB, Dredge K, Dalgleish AG. The evolution of thalidomide and its IMiD derivatives as anticancer agents. Nat Rev Cancer. 2004;4: 314-322. - PubMed
-
- Teo SK, Resztak KE, Scheffler MA, et al. Thalidomide in the treatment of leprosy. Microbes Infect. 2002;4: 1193-1202. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
