

LPS-induced TNF-alpha factor (LITAF)-deficient mice express reduced LPS-induced cytokine: Evidence for LITAF-dependent LPS signaling pathways
- PMID: 16954198
- PMCID: PMC1560089
- DOI: 10.1073/pnas.0605988103
LPS-induced TNF-alpha factor (LITAF)-deficient mice express reduced LPS-induced cytokine: Evidence for LITAF-dependent LPS signaling pathways
Erratum in
- Proc Natl Acad Sci U S A. 2007 Feb 20;104(8):3015
Abstract
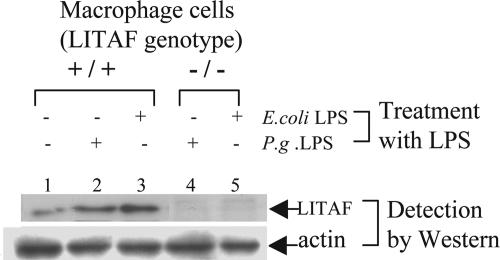
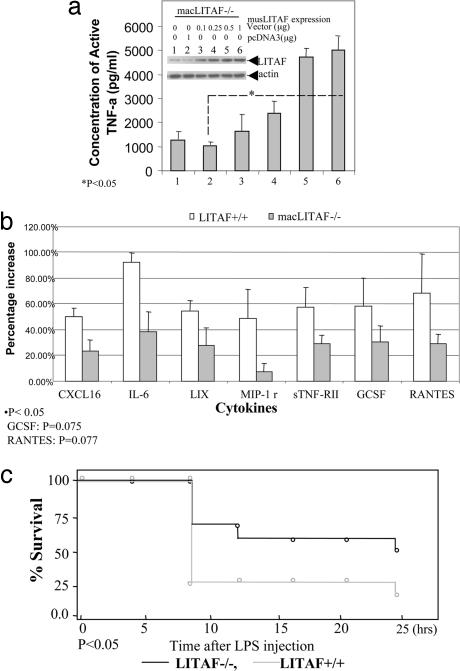
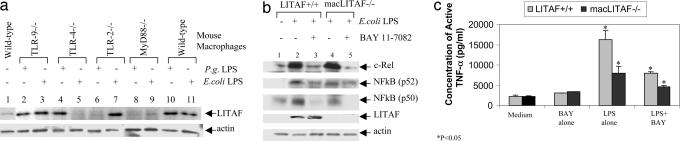
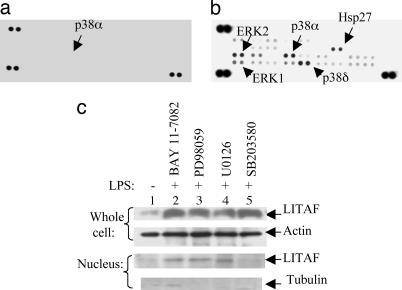
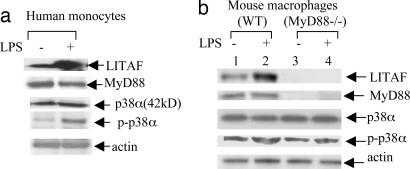
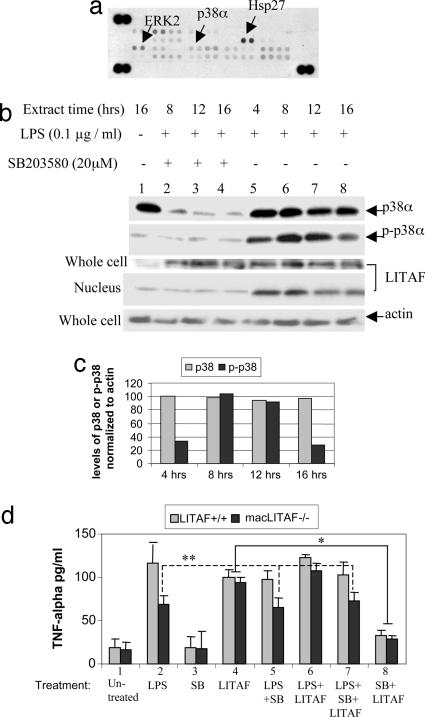
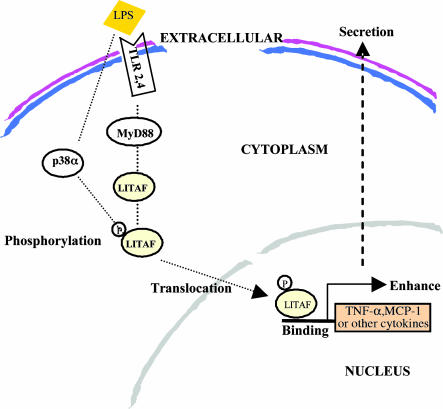
Previously we identified a transcription factor, LPS-Induced TNF-alpha Factor (LITAF), mediating inflammatory cytokine expression in LPS-induced processes. To characterize the role of LITAF in vivo, we generated a macrophage-specific LITAF-deficient mouse (macLITAF(-/-)). Our data demonstrate that in macrophages (i) several cytokines (such as TNF-alpha, IL-6, sTNF-RII, and CXCL16) are induced at lower levels in macLITAF(-/-) compared with LITAF(+/+) control macrophages; (ii) macLITAF(-/-) mice are more resistant to LPS-induced lethality. To further identify LITAF signaling pathways, we tested mouse TLR-2(-/-), -4(-/-), and -9(-/-) and WT peritoneal macrophages exposed to LPS. Using these cells, we now show that LITAF expression can be induced after challenge either with LPS from Porphyromonas gingivalis via agonism at TLR-2, or with LPS from Escherichia coli via agonism at TLR-4, both requiring functional MyD88. We also show that, in response to LPS, the MyD88-dependent LITAF pathway differs from the NF-kappaB pathway. Furthermore, using a kinase array, p38alpha was found to mediate LITAF phosphorylation and the inhibition of p38alpha with a p38-specific inhibitor (SB203580) blocked LITAF nuclear translocation and reduced LPS-induced TNF-alpha protein levels. Finally, macLITAF(-/-) macrophages rescued by LITAF cDNA transfection restored levels of TNF-alpha similar to those observed in WT cells. We conclude that LITAF is an important mediator of the LPS-induced inflammatory response that can be distinguished from NF-kappaB pathway and that p38alpha is the specific kinase involved in the pathway linking LPS/MyD88/LITAF to TNF.
Conflict of interest statement
Conflict of interest statement: No conflicts declared.
Figures
Similar articles
-
Whole-body deletion of LPS-induced TNF-α factor (LITAF) markedly improves experimental endotoxic shock and inflammatory arthritis.Proc Natl Acad Sci U S A. 2011 Dec 27;108(52):21247-52. doi: 10.1073/pnas.1111492108. Epub 2011 Dec 12. Proc Natl Acad Sci U S A. 2011. PMID: 22160695 Free PMC article.
-
Dynamic regulation of pro- and anti-inflammatory cytokines by MAPK phosphatase 1 (MKP-1) in innate immune responses.Proc Natl Acad Sci U S A. 2006 Feb 14;103(7):2274-9. doi: 10.1073/pnas.0510965103. Epub 2006 Feb 6. Proc Natl Acad Sci U S A. 2006. PMID: 16461893 Free PMC article.
-
Induction of in vitro reprogramming by Toll-like receptor (TLR)2 and TLR4 agonists in murine macrophages: effects of TLR "homotolerance" versus "heterotolerance" on NF-kappa B signaling pathway components.J Immunol. 2003 Jan 1;170(1):508-19. doi: 10.4049/jimmunol.170.1.508. J Immunol. 2003. PMID: 12496438
-
β(2) integrins inhibit TLR responses by regulating NF-κB pathway and p38 MAPK activation.Eur J Immunol. 2013 Mar;43(3):779-92. doi: 10.1002/eji.201242550. Epub 2013 Feb 11. Eur J Immunol. 2013. PMID: 23310953 Free PMC article.
-
Kavain Involvement in LPS-Induced Signaling Pathways.J Cell Biochem. 2016 Oct;117(10):2272-80. doi: 10.1002/jcb.25525. Epub 2016 Mar 9. J Cell Biochem. 2016. PMID: 26917453
Cited by
-
SIMPLE: A new regulator of endosomal trafficking and signaling in health and disease.Commun Integr Biol. 2013 May 1;6(3):e24214. doi: 10.4161/cib.24214. Epub 2013 Apr 9. Commun Integr Biol. 2013. PMID: 23713142 Free PMC article.
-
LITAF mediation of increased TNF-α secretion from inflamed colonic lamina propria macrophages.PLoS One. 2011;6(9):e25849. doi: 10.1371/journal.pone.0025849. Epub 2011 Sep 30. PLoS One. 2011. PMID: 21984950 Free PMC article.
-
Molecular cloning of adipose triglyceride lipase (ATGL) gene from blunt snout bream and its expression after LPS-induced TNF-α factor.Fish Physiol Biochem. 2018 Aug;44(4):1143-1157. doi: 10.1007/s10695-018-0502-4. Epub 2018 Apr 29. Fish Physiol Biochem. 2018. PMID: 29705966
-
LINC00173 silence and estrone supply suppress ER+ breast cancer by estrogen receptor α degradation and LITAF activation.Cancer Sci. 2024 Jul;115(7):2318-2332. doi: 10.1111/cas.16201. Epub 2024 May 5. Cancer Sci. 2024. PMID: 38705575 Free PMC article.
-
Induction of TNF-alpha by LPS in Schwann cell is regulated by MAPK activation signals.Cell Mol Neurobiol. 2007 Nov;27(7):909-21. doi: 10.1007/s10571-007-9215-4. Epub 2007 Sep 28. Cell Mol Neurobiol. 2007. PMID: 17902045 Free PMC article.
References
-
- Beutler B, Hoebe K, Du X, Ulevitch RJ. J Leukoc Biol. 2003;74:479–485. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
