

Searching for IRES
- PMID: 16957278
- PMCID: PMC1581980
- DOI: 10.1261/rna.157806
Searching for IRES
Abstract
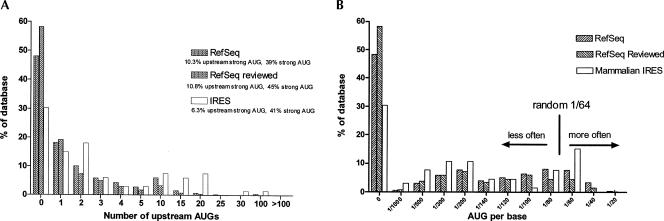
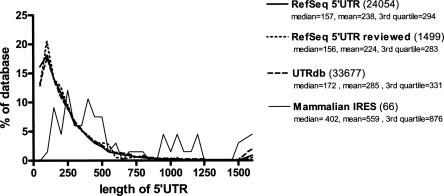
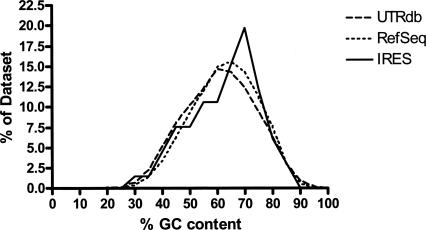
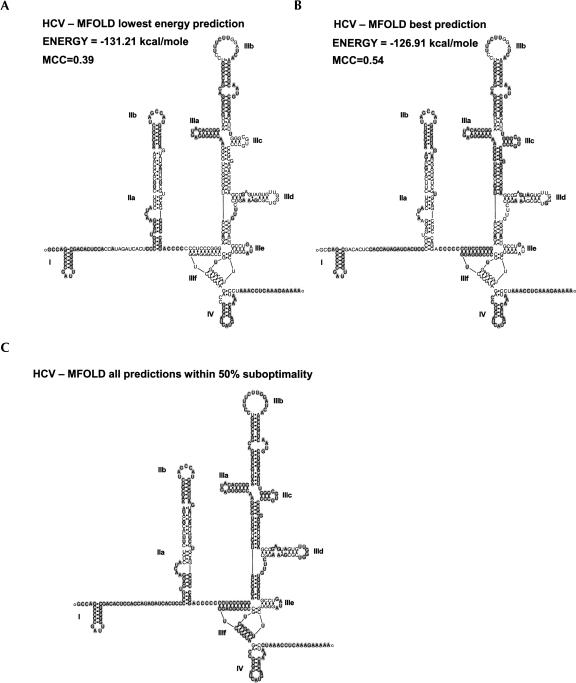
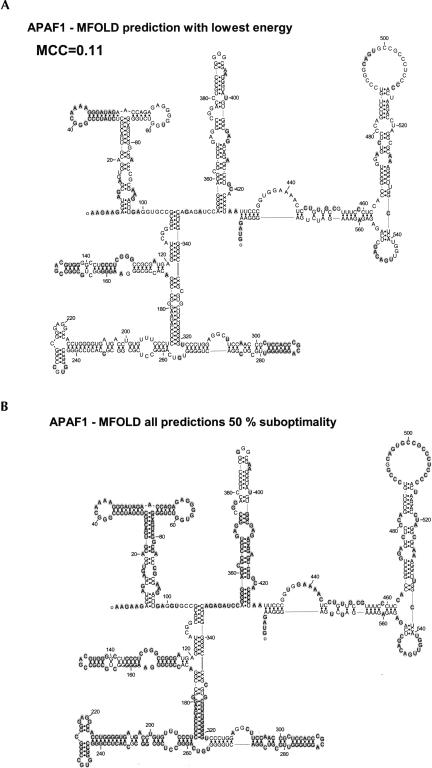
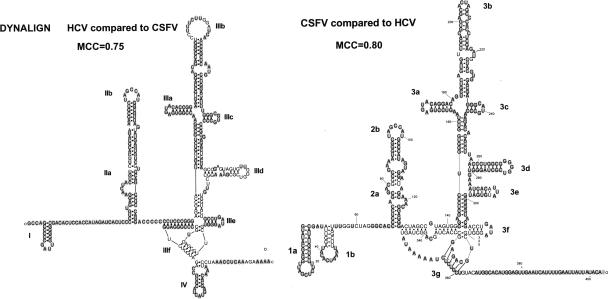
The cell has many ways to regulate the production of proteins. One mechanism is through the changes to the machinery of translation initiation. These alterations favor the translation of one subset of mRNAs over another. It was first shown that internal ribosome entry sites (IRESes) within viral RNA genomes allowed the production of viral proteins more efficiently than most of the host proteins. The RNA secondary structure of viral IRESes has sometimes been conserved between viral species even though the primary sequences differ. These structures are important for IRES function, but no similar structure conservation has yet to be shown in cellular IRES. With the advances in mathematical modeling and computational approaches to complex biological problems, is there a way to predict an IRES in a data set of unknown sequences? This review examines what is known about cellular IRES structures, as well as the data sets and tools available to examine this question. We find that the lengths, number of upstream AUGs, and %GC content of 5'-UTRs of the human transcriptome have a similar distribution to those of published IRES-containing UTRs. Although the UTRs containing IRESes are on the average longer, almost half of all 5'-UTRs are long enough to contain an IRES. Examination of the available RNA structure prediction software and RNA motif searching programs indicates that while these programs are useful tools to fine tune the empirically determined RNA secondary structure, the accuracy of de novo secondary structure prediction of large RNA molecules and subsequent identification of new IRES elements by computational approaches, is still not possible.
Figures

References
-
- Akbergenov, R., Zhanybekova, S., Kryldakov, R.V., Zhigailov, A., Polimbetova, N.S., Hohn, T., Iskakov, B.K. ARC-1, a sequence element complementary to an internal 18S rRNA segment, enhances translation efficiency in plants when present in the leader or intercistronic region of mRNAs. Nucleic Acids Res. 2004;32:239–247. - PMC - PubMed
-
- Anwar, A., Ali, N., Tanveer, R., Siddiqui, A. Demonstration of functional requirement of polypyrimidine tract-binding protein by SELEX RNA during hepatitis C virus internal ribosome entry site-mediated translation initiation. J. Biol. Chem. 2000;275:34231–34235. - PubMed
-
- Attal, J., Theron, M.C., Taboit, F., Cajero-Juarez, M., Kann, G., Bolifraud, P., Houdebine, L.M. The RU5 (‘R’) region from human leukaemia viruses (HTLV-1) contains an internal ribosome entry site (IRES)-like sequence. FEBS Lett. 1996;392:220–224. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous