

A rapid, reversible, and tunable method to regulate protein function in living cells using synthetic small molecules
- PMID: 16959577
- PMCID: PMC3290523
- DOI: 10.1016/j.cell.2006.07.025
A rapid, reversible, and tunable method to regulate protein function in living cells using synthetic small molecules
Abstract
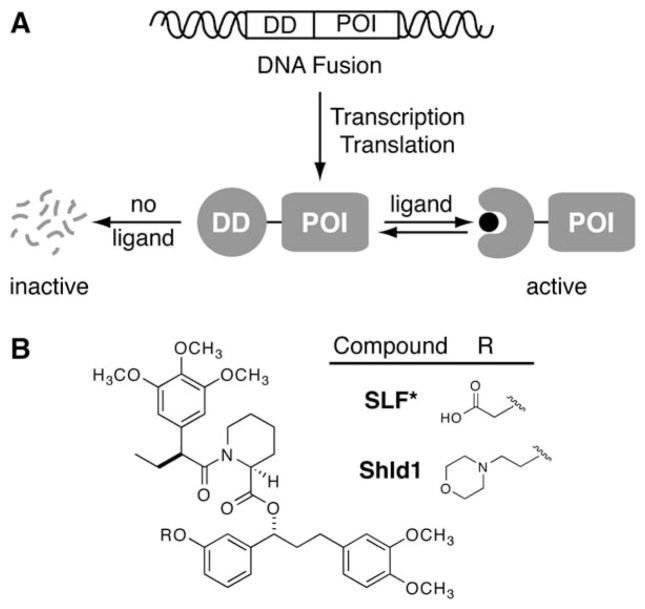
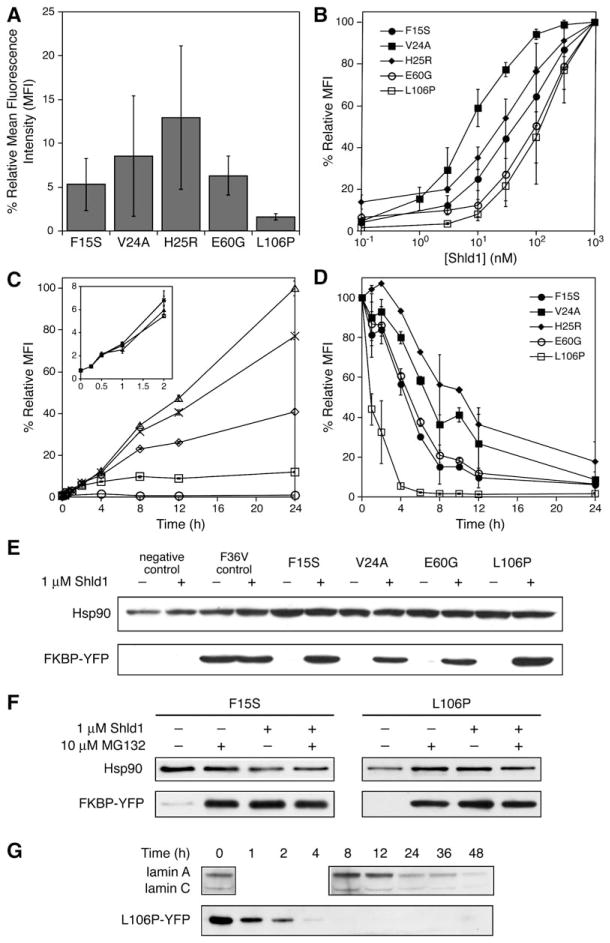
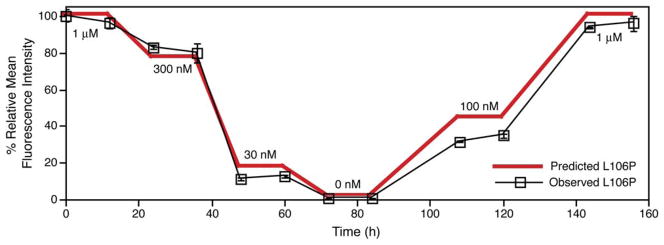
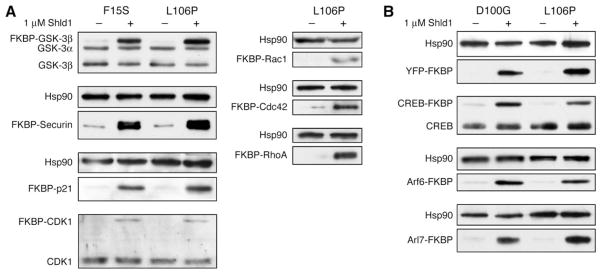
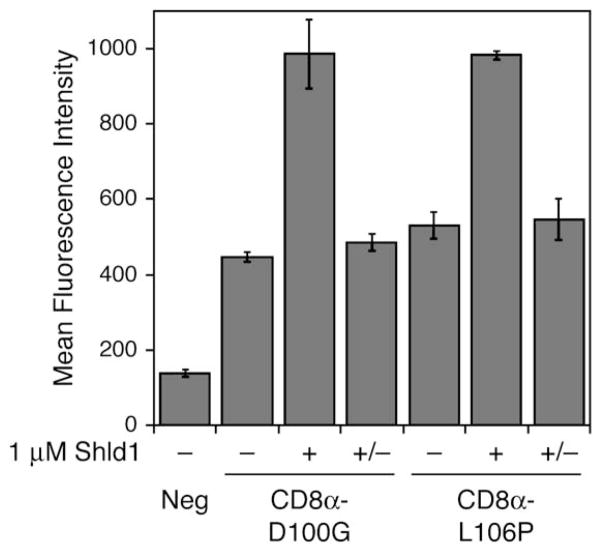
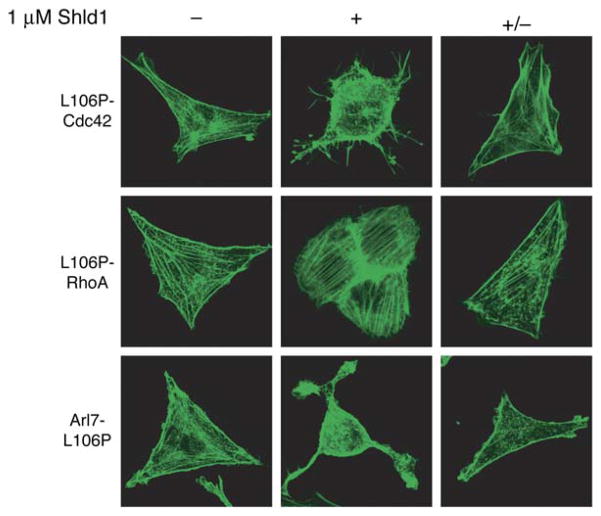
Rapid and reversible methods for perturbing the function of specific proteins are desirable tools for probing complex biological systems. We have developed a general technique to regulate the stability of specific proteins in mammalian cells using cell-permeable, synthetic molecules. We engineered mutants of the human FKBP12 protein that are rapidly and constitutively degraded when expressed in mammalian cells, and this instability is conferred to other proteins fused to these destabilizing domains. Addition of a synthetic ligand that binds to the destabilizing domains shields them from degradation, allowing fused proteins to perform their cellular functions. Genetic fusion of the destabilizing domain to a gene of interest ensures specificity, and the attendant small-molecule control confers speed, reversibility, and dose-dependence to this method. This general strategy for regulating protein stability should enable conditional perturbation of specific proteins with unprecedented control in a variety of experimental settings.
Figures
Comment in
-
Targeting protein stability with a small molecule.Cell. 2006 Sep 8;126(5):827-9. doi: 10.1016/j.cell.2006.08.023. Cell. 2006. PMID: 16959558
References
-
- Bachmair A, Finley D, Varshavsky A. In vivo half-life of a protein is a function of its amino-terminal residue. Science. 1986;234:179–186. - PubMed
-
- Banaszynski LA, Wandless TJ. Conditional control of protein function. Chem Biol. 2006;13:11–21. - PubMed
-
- Bence NF, Sampat RM, Kopito RR. Impairment of the ubiquitin-proteasome system by protein aggregation. Science. 2001;292:1552–1555. - PubMed
-
- Bishop AC, Shah K, Liu Y, Witucki L, Kung CY, Shokat KM. Design of allele-specific inhibitors to probe protein kinase signaling. Curr Biol. 1998;8:257–266. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
