

Disulfide isomerization switches tissue factor from coagulation to cell signaling
- PMID: 16959886
- PMCID: PMC1599891
- DOI: 10.1073/pnas.0606411103
Disulfide isomerization switches tissue factor from coagulation to cell signaling
Abstract
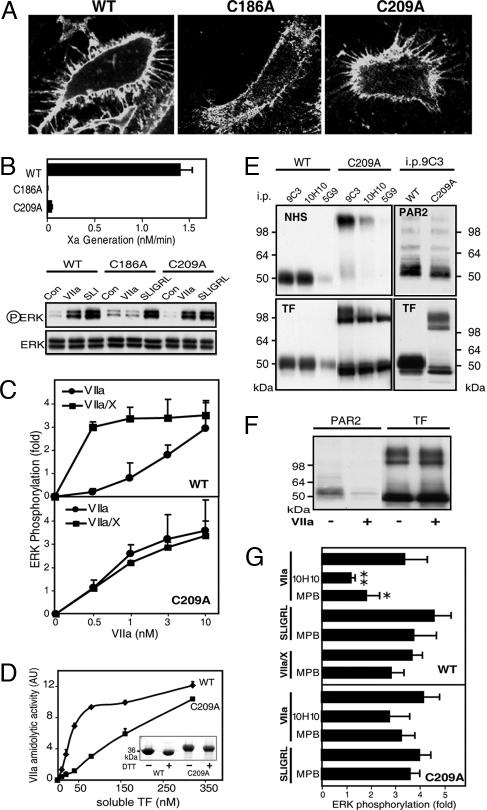
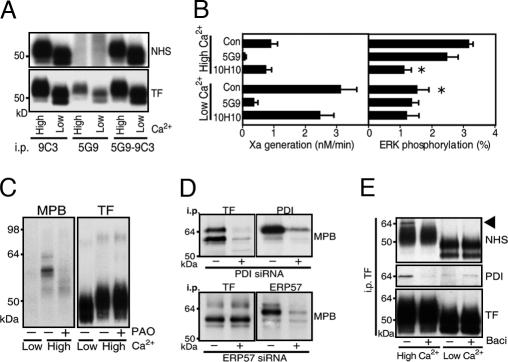
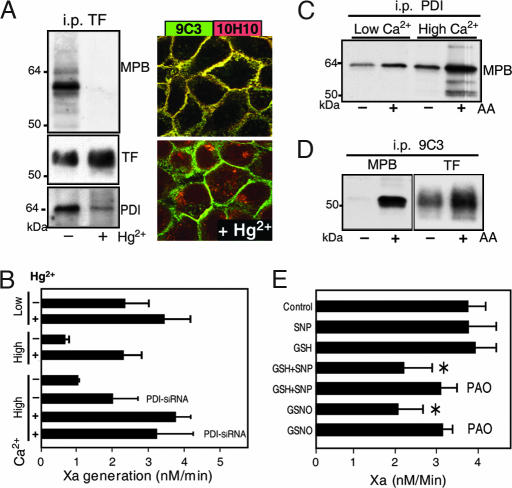
Cell-surface tissue factor (TF) binds the serine protease factor VIIa to activate coagulation or, alternatively, to trigger signaling through the G protein-coupled, protease-activated receptor 2 (PAR2) relevant to inflammation and angiogenesis. Here we demonstrate that TF.VIIa-mediated coagulation and cell signaling involve distinct cellular pools of TF. The surface-accessible, extracellular Cys186-Cys209 disulfide bond of TF is critical for coagulation, and protein disulfide isomerase (PDI) disables coagulation by targeting this disulfide. A TF mutant (TF C209A) with an unpaired Cys186 retains TF.VIIa signaling activity, and it has reduced affinity for VIIa, a characteristic of signaling TF on cells with constitutive TF expression. We further show that PDI suppresses TF coagulant activity in a nitric oxide-dependent pathway, linking the regulation of TF thrombogenicity to oxidative stress in the vasculature. Furthermore, a unique monoclonal antibody recognizes only the noncoagulant, cryptic conformation of TF. This antibody inhibits formation of the TF.PAR2 complex and TF.VIIa signaling, but it does not prevent coagulation activation. These experiments delineate an upstream regulatory mechanism that controls TF function, and they provide initial evidence that TF.VIIa signaling can be specifically inhibited with minimal effects on coagulation.
Conflict of interest statement
Conflict of interest statement: No conflicts declared.
Figures


Comment in
-
Cryptic messages: is noncoagulant tissue factor reserved for cell signaling?Proc Natl Acad Sci U S A. 2006 Sep 26;103(39):14259-60. doi: 10.1073/pnas.0606888103. Epub 2006 Sep 19. Proc Natl Acad Sci U S A. 2006. PMID: 16985001 Free PMC article. No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
