

A deleterious mutation in SAMD9 causes normophosphatemic familial tumoral calcinosis
- PMID: 16960814
- PMCID: PMC1592555
- DOI: 10.1086/508069
A deleterious mutation in SAMD9 causes normophosphatemic familial tumoral calcinosis
Abstract
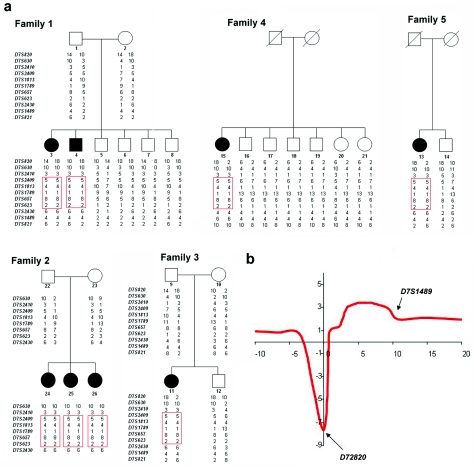
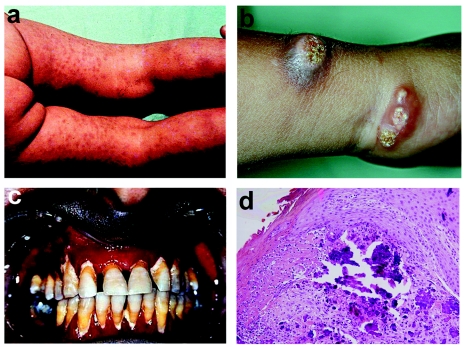
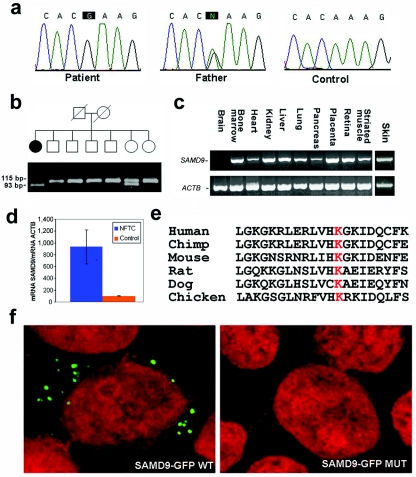
Familial tumoral calcinosis (FTC) is a rare autosomal recessive disorder characterized by the progressive deposition of calcified masses in cutaneous and subcutaneous tissues, which results in painful ulcerative lesions and severe skin and bone infections. Two major types of FTC have been recognized: hyperphosphatemic FTC (HFTC) and normophosphatemic FTC (NFTC). HFTC was recently shown to result from mutations in two different genes: GALNT3, which codes for a glycosyltransferase, and FGF23, which codes for a potent phosphaturic protein. To determine the molecular cause of NFTC, we performed homozygosity mapping in five affected families of Jewish Yemenite origin and mapped NFTC to 7q21-7q21.3. Mutation analysis revealed a homozygous mutation in the SAMD9 gene (K1495E), which was found to segregate with the disease in all families and to interfere with the protein expression. Our data suggest that SAMD9 is involved in the regulation of extraosseous calcification, a process of considerable importance in a wide range of diseases as common as atherosclerosis and autoimmune disorders.
Figures
References
Web Resources
-
- Genetic Linkage Analysis Project, http://cbl-fog.cs.technion.ac.il/superlink/ (for Superlink online software)
-
- Map Viewer, http://www.ncbi.nlm.nih.gov/mapview/
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for HFTC and hyperostosis-hyperphosphatemia syndrome)
References
-
- Atzeni F, Sarzi-Puttini P, Bevilacqua M (2006) Calcium deposition and associated chronic diseases (atherosclerosis, diffuse idiopathic skeletal hyperostosis, and others). Rheum Dis Clin North Am 32:413–426 - PubMed
-
- White KE, Larsson TE, Econs MJ (2006) The roles of specific genes implicated as circulating factors involved in normal and disordered phosphate homeostasis: frizzled related protein-4, matrix extracellular phosphoglycoprotein, and fibroblast growth factor 23. Endocr Rev 27:221–24110.1210/er.2005-0019 - DOI - PubMed
-
- Topaz O, Shurman DL, Bergman R, Indelman M, Ratajczak P, Mizrachi M, Khamaysi Z, Behar D, Petronius D, Friedman V, Zelikovic I, Raimer S, Metzker A, Richard G, Sprecher E (2004) Mutations in GALNT3, encoding a protein involved in O-linked glycosylation, cause familial tumoral calcinosis. Nat Genet 36:579–58110.1038/ng1358 - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
