

Preconditioning: a paradigm shift in the biology of myocardial ischemia
- PMID: 16963615
- PMCID: PMC3242363
- DOI: 10.1152/ajpheart.00712.2006
Preconditioning: a paradigm shift in the biology of myocardial ischemia
Abstract
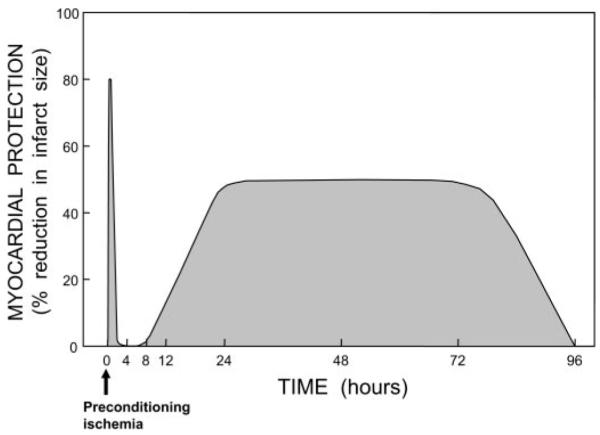
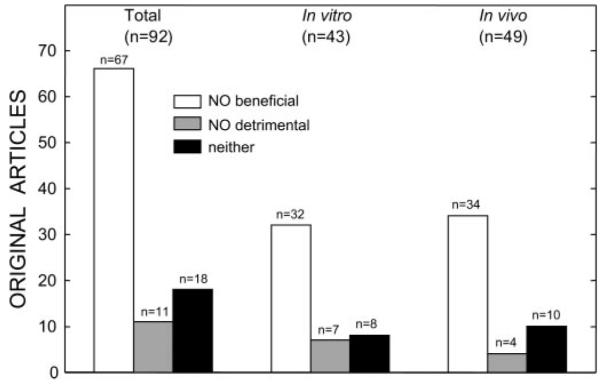
The discovery of preconditioning (PC) has arguably been the single most important development in the field of ischemic biology in the past 20 years. The significance of this phenomenon transcends cardiovascular medicine, since it is ubiquitously observed in virtually every tissue of the body. This article reviews the pathophysiology and molecular basis of myocardial PC, with particular emphasis on the late phase of this cardioprotective adaptation. The article also discusses the exploitation of late PC for the development of novel gene therapy strategies aimed at inducing a permanently preconditioned cardiac phenotype (prophylactic cardioprotection). Besides its conceptual interest, deciphering the mechanism of late PC has considerable therapeutic reverberations, since transfer of the genes that underlie late PC would be expected to emulate the salubrious effects of this response of the heart to stress.
Figures



References
-
- Abete P, Ferrara N, Cioppa A, Ferrara P, Bianco S, Calabrese C, Cacciatore F, Longobardi G, Rengo F. Preconditioning does not prevent postischemic dysfunction in aging heart. J Am Coll Cardiol. 1996;27:1777–1786. - PubMed
-
- Bolli R. Cardioprotective function of inducible nitric oxide synthase and role of nitric oxide in myocardial ischemia and preconditioning: an overview of a decade of research. J Mol Cell Cardiol. 2001;33:1897–1918. - PubMed
-
- Bolli R. The late phase of preconditioning. Circ Res. 2000;87:972–983. - PubMed
-
- Bolli R, Becker L, Gross G, Mentzer R, Jr, Balshaw D, Lathrop DA. Myocardial protection at a crossroads: the need for translation into clinical therapy. Circ Res. 2004;95:125–134. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous