

Comparative genomics modeling of the NRSF/REST repressor network: from single conserved sites to genome-wide repertoire
- PMID: 16963704
- PMCID: PMC1581430
- DOI: 10.1101/gr.4997306
Comparative genomics modeling of the NRSF/REST repressor network: from single conserved sites to genome-wide repertoire
Abstract
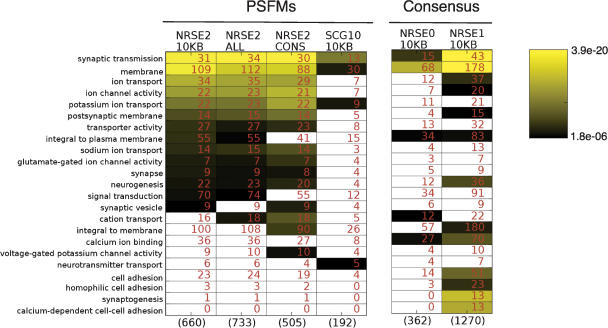
We constructed and applied an open source informatic framework called Cistematic in an effort to predict the target gene repertoire for transcription factors with large binding sites. Cistematic uses two different evolutionary conservation-filtering algorithms in conjunction with several analysis modules. Beginning with a single conserved and biologically tested site for the neuronal repressor NRSF/REST, Cistematic generated a refined PSFM (position specific frequency matrix) based on conserved site occurrences in mouse, human, and dog genomes. Predictions from this model were validated by chromatin immunoprecipitation (ChIP) followed by quantitative PCR. The combination of transfection assays and ChIP enrichment data provided an objective basis for setting a threshold for membership and rank-ordering a final gene cohort model consisting of 842 high-confidence sites in the human genome associated with 733 genes. Statistically significant enrichment of NRSE-associated genes was found for neuron-specific Gene Ontology (GO) terms and neuronal mRNA expression profiles. A more extensive evolutionary survey showed that NRSE sites matching the PSFM model exist in roughly similar numbers in all fully sequenced vertebrate genomes but are notably absent from invertebrate and protochordate genomes, as is NRSF itself. Some NRSF/REST sites reside in repeats, which suggests a mechanism for both ancient and modern dispersal of NRSEs through vertebrate genomes. Multiple predicted sites are located near neuronal microRNA and splicing-factor genes, and these tested positive for NRSF/REST occupancy in vivo. The resulting network model integrates post-transcriptional and translational controllers, including candidate feedback loops on NRSF and its corepressor, CoREST.
Figures







References
-
- Abderrahmani A., Steinmann M., Plaisance V., Niederhauser G., Haefliger J.A., Mooser V., Bonny C., Nicod P., Waeber G., Steinmann M., Plaisance V., Niederhauser G., Haefliger J.A., Mooser V., Bonny C., Nicod P., Waeber G., Plaisance V., Niederhauser G., Haefliger J.A., Mooser V., Bonny C., Nicod P., Waeber G., Niederhauser G., Haefliger J.A., Mooser V., Bonny C., Nicod P., Waeber G., Haefliger J.A., Mooser V., Bonny C., Nicod P., Waeber G., Mooser V., Bonny C., Nicod P., Waeber G., Bonny C., Nicod P., Waeber G., Nicod P., Waeber G., Waeber G. The transcriptional repressor REST determines the cell-specific expression of the human MAPK8IP1 gene encoding IB1 (JIP-1) Mol. Cell. Biol. 2001;21:7256–7267. - PMC - PubMed
-
- Andres M.E., Burger C., Peral-Rubio M.J., Battaglioli E., Anderson M.E., Grimes J., Dallman J., Ballas N., Mandel G., Burger C., Peral-Rubio M.J., Battaglioli E., Anderson M.E., Grimes J., Dallman J., Ballas N., Mandel G., Peral-Rubio M.J., Battaglioli E., Anderson M.E., Grimes J., Dallman J., Ballas N., Mandel G., Battaglioli E., Anderson M.E., Grimes J., Dallman J., Ballas N., Mandel G., Anderson M.E., Grimes J., Dallman J., Ballas N., Mandel G., Grimes J., Dallman J., Ballas N., Mandel G., Dallman J., Ballas N., Mandel G., Ballas N., Mandel G., Mandel G. CoREST: A functional corepressor required for regulation of neural-specific gene expression. Proc. Natl. Acad. Sci. 1999;96:9873–9878. - PMC - PubMed
-
- Atouf F., Czernichow P., Scharfmann R., Czernichow P., Scharfmann R., Scharfmann R. Expression of neuronal traits in pancreatic β cells—Implication ofneuron-restrictive silencing factor/repressor element silencing transcription factor, a neuron-restrictive silencer. J. Biol. Chem. 1997;272:1929–1934. - PubMed
-
- Ballas N., Grunseich C., Lu D.D., Speh J.C., Mandel G., Grunseich C., Lu D.D., Speh J.C., Mandel G., Lu D.D., Speh J.C., Mandel G., Speh J.C., Mandel G., Mandel G. REST and its corepressors mediate plasticity of neuronal gene chromatin throughout neurogenesis. Cell. 2005;121:645–657. - PubMed
-
- Boffelli D., Weer C.V., Weng L., Lewis K.D., Shoukry M.I., Pachter L., Keys D.N., Rubin E.M., Weer C.V., Weng L., Lewis K.D., Shoukry M.I., Pachter L., Keys D.N., Rubin E.M., Weng L., Lewis K.D., Shoukry M.I., Pachter L., Keys D.N., Rubin E.M., Lewis K.D., Shoukry M.I., Pachter L., Keys D.N., Rubin E.M., Shoukry M.I., Pachter L., Keys D.N., Rubin E.M., Pachter L., Keys D.N., Rubin E.M., Keys D.N., Rubin E.M., Rubin E.M. Intraspecies sequence comparisons for annotating genomes. Genome Res. 2004;14:2406–2411. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources