

beta-Arrestin2 mediates nephrin endocytosis and impairs slit diaphragm integrity
- PMID: 16968782
- PMCID: PMC1564064
- DOI: 10.1073/pnas.0602587103
beta-Arrestin2 mediates nephrin endocytosis and impairs slit diaphragm integrity
Abstract
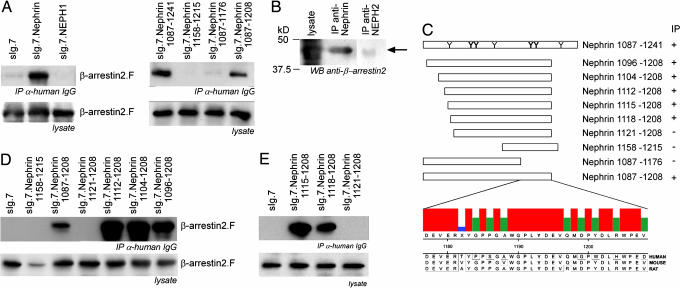
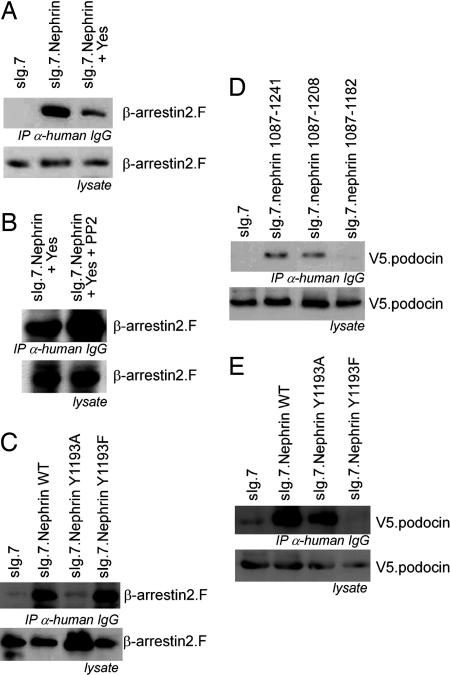
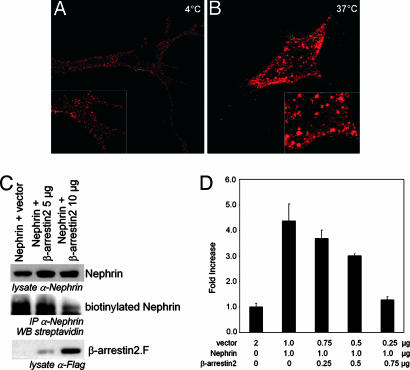
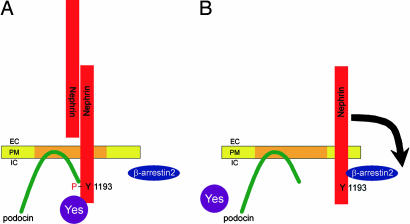
beta-Arrestins mediate internalization of plasma membrane receptors. Nephrin, a structural component of the glomerular slit diaphragm, is a single transmembrane spanning receptor and belongs to the family of adhesion molecules. Its mutation causes a hereditary nephrotic syndrome. We report the previously undescribed interaction of beta-arrestin2 with the nephrin C terminus. The phosphorylation status of nephrin Y1193 regulates inversely the binding of beta-arrestin2 and podocin. The Src-family member Yes, known to enhance podocin-nephrin interaction by nephrin phosphorylation, diminishes beta-arrestin2-nephrin interaction. beta-Arrestin2 induces nephrin endocytosis and attenuates nephrin signaling. This finding suggests that nephrin Y1193 serves as a molecular switch that determines the integrity of the slit diaphragm by functional competition between beta-arrestin2 and podocin. This concept offers a molecular pathomechanism of slit diaphragm distortion and opens therapeutic avenues for glomerular diseases.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Kestila M, Lenkkeri U, Mannikko M, Lamerdin J, McCready P, Putaala H, Ruotsalainen V, Morita T, Nissinen M, Herva R, et al. Mol Cell. 1998;1:575–582. - PubMed
-
- Tryggvason K, Patrakka J, Wartiovaara J. N Engl J Med. 2006;354:1387–1401. - PubMed
-
- Peterson JC, Adler S, Burkart JM, Greene T, Hebert LA, Hunsicker LG, King AJ, Klahr S, Massry SG, Seifter JL. Ann Intern Med. 1995;123:754–762. - PubMed
-
- Shih NY, Li J, Karpitskii V, Nguyen A, Dustin ML, Kanagawa O, Miner JH, Shaw AS. Science. 1999;286:312–315. - PubMed
-
- Boute N, Gribouval O, Roselli S, Benessy F, Lee H, Fuchshuber A, Dahan K, Gubler MC, Niaudet P, Antignac C. Nat Genet. 2000;24:349–354. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
