

Application of protein structure alignments to iterated hidden Markov model protocols for structure prediction
- PMID: 16970830
- PMCID: PMC1622756
- DOI: 10.1186/1471-2105-7-410
Application of protein structure alignments to iterated hidden Markov model protocols for structure prediction
Abstract
Background: One of the most powerful methods for the prediction of protein structure from sequence information alone is the iterative construction of profile-type models. Because profiles are built from sequence alignments, the sequences included in the alignment and the method used to align them will be important to the sensitivity of the resulting profile. The inclusion of highly diverse sequences will presumably produce a more powerful profile, but distantly related sequences can be difficult to align accurately using only sequence information. Therefore, it would be expected that the use of protein structure alignments to improve the selection and alignment of diverse sequence homologs might yield improved profiles. However, the actual utility of such an approach has remained unclear.
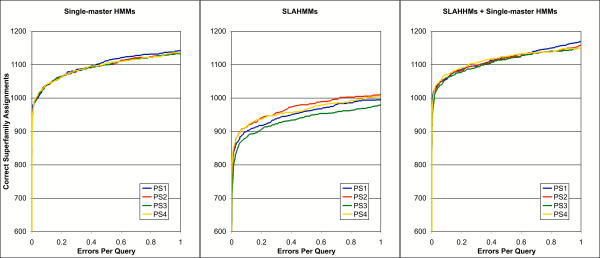
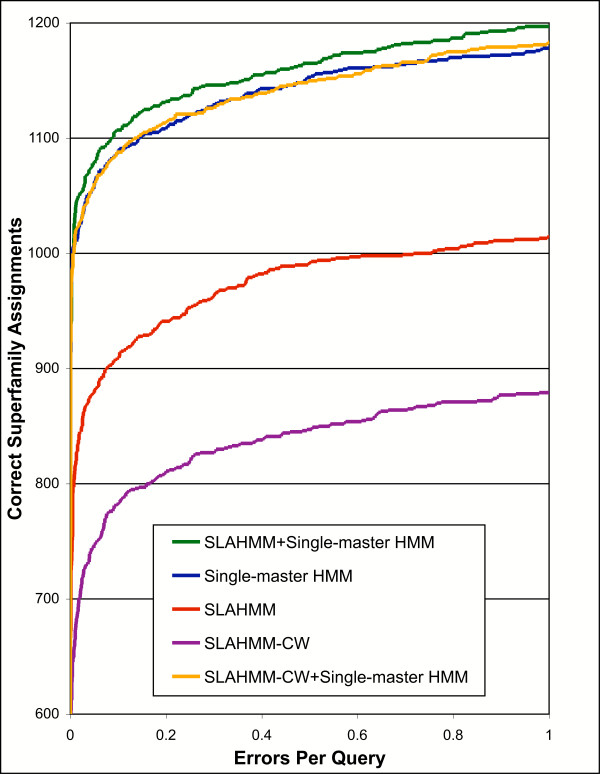
Results: We explored several iterative protocols for the generation of profile hidden Markov models. These protocols were tailored to allow the inclusion of protein structure alignments in the process, and were used for large-scale creation and benchmarking of structure alignment-enhanced models. We found that models using structure alignments did not provide an overall improvement over sequence-only models for superfamily-level structure predictions. However, the results also revealed that the structure alignment-enhanced models were complimentary to the sequence-only models, particularly at the edge of the "twilight zone". When the two sets of models were combined, they provided improved results over sequence-only models alone. In addition, we found that the beneficial effects of the structure alignment-enhanced models could not be realized if the structure-based alignments were replaced with sequence-based alignments. Our experiments with different iterative protocols for sequence-only models also suggested that simple protocol modifications were unable to yield equivalent improvements to those provided by the structure alignment-enhanced models. Finally, we found that models using structure alignments provided fold-level structure assignments that were superior to those produced by sequence-only models.
Conclusion: When attempting to predict the structure of remote homologs, we advocate a combined approach in which both traditional models and models incorporating structure alignments are used.
Figures



Similar articles
-
HMM-ModE--improved classification using profile hidden Markov models by optimising the discrimination threshold and modifying emission probabilities with negative training sequences.BMC Bioinformatics. 2007 Mar 27;8:104. doi: 10.1186/1471-2105-8-104. BMC Bioinformatics. 2007. PMID: 17389042 Free PMC article.
-
Non-sequential structure-based alignments reveal topology-independent core packing arrangements in proteins.Bioinformatics. 2005 Apr 1;21(7):1010-9. doi: 10.1093/bioinformatics/bti128. Epub 2004 Nov 5. Bioinformatics. 2005. PMID: 15531601
-
Quasi-consensus-based comparison of profile hidden Markov models for protein sequences.Bioinformatics. 2005 May 15;21(10):2287-93. doi: 10.1093/bioinformatics/bti374. Epub 2005 Mar 29. Bioinformatics. 2005. PMID: 15797916
-
Sequence comparison and protein structure prediction.Curr Opin Struct Biol. 2006 Jun;16(3):374-84. doi: 10.1016/j.sbi.2006.05.006. Epub 2006 May 19. Curr Opin Struct Biol. 2006. PMID: 16713709 Review.
-
An introduction to modeling structure from sequence.Curr Protoc Bioinformatics. 2006 Oct;Chapter 5:Unit 5.1. doi: 10.1002/0471250953.bi0501s15. Curr Protoc Bioinformatics. 2006. PMID: 18428765 Review.
Cited by
-
Refining homology models by combining replica-exchange molecular dynamics and statistical potentials.Proteins. 2008 Sep;72(4):1171-88. doi: 10.1002/prot.22005. Proteins. 2008. PMID: 18338384 Free PMC article.
-
Comprehensive prediction of chromosome dimer resolution sites in bacterial genomes.BMC Genomics. 2011 Jan 11;12:19. doi: 10.1186/1471-2164-12-19. BMC Genomics. 2011. PMID: 21223577 Free PMC article.
-
Hidden Markov model speed heuristic and iterative HMM search procedure.BMC Bioinformatics. 2010 Aug 18;11:431. doi: 10.1186/1471-2105-11-431. BMC Bioinformatics. 2010. PMID: 20718988 Free PMC article.
-
Detection and architecture of small heat shock protein monomers.PLoS One. 2010 Apr 7;5(4):e9990. doi: 10.1371/journal.pone.0009990. PLoS One. 2010. PMID: 20383329 Free PMC article.
References
-
- Aloy P, Querol E, Aviles FX, Sternberg MJ. Automated structure-based prediction of functional sites in proteins: applications to assessing the validity of inheriting protein function from homology in genome annotation and to protein docking. J Mol Biol. 2001;311:395–408. doi: 10.1006/jmbi.2001.4870. - DOI - PubMed
