

A high-throughput method for cloning and sequencing human immunodeficiency virus type 1 integration sites
- PMID: 16971446
- PMCID: PMC1642152
- DOI: 10.1128/JVI.01737-06
A high-throughput method for cloning and sequencing human immunodeficiency virus type 1 integration sites
Abstract
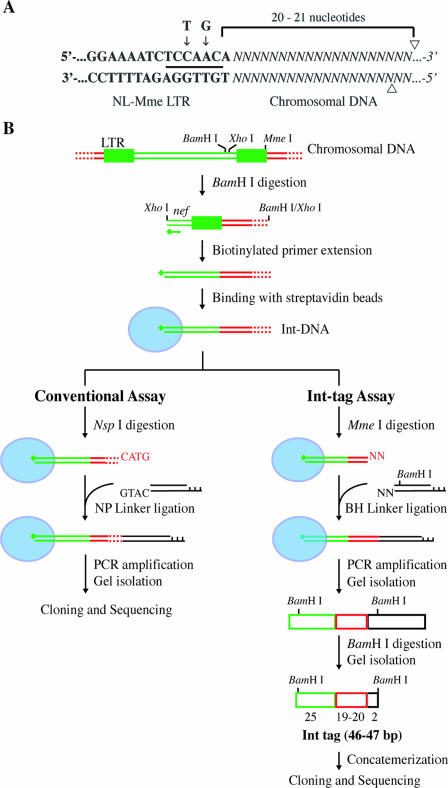
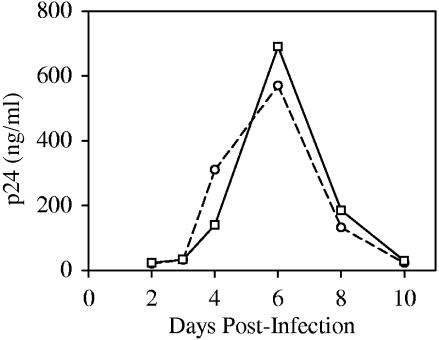
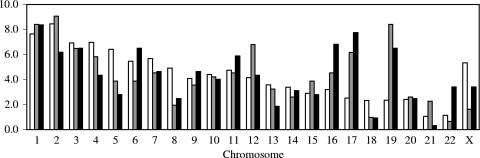
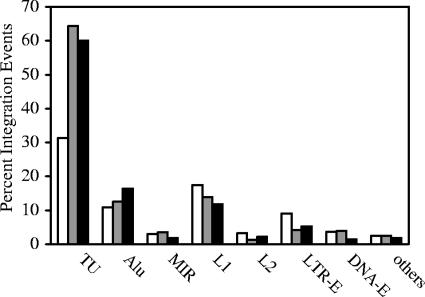
Integration of retroviral DNA is nonspecific and can occur at many sites throughout chromosomes. However, the process is not uniformly distributed, and both hot and cold spots for integration exist. The mechanism that determines target site specificity is not well understood. Because of the nonspecific and widespread nature of integration, studies analyzing the mechanism and factors that control target site selection require the collection and analysis of a large library of human immunodeficiency virus type 1 (HIV-1) proviral clones. Such analyses are time-consuming and labor-intensive using conventional means. We have developed an efficient and high-throughput method of sequencing and mapping a large number of independent integration sites in the absence of any selection or bias. The new assay involves the use of a modified HIV-1 (NL-Mme) containing a type IIS restriction site, MmeI, at the right end of viral DNA. Digestion of genomic DNA from NL-Mme-infected cells generated viral DNA-containing fragments of a discrete size. Subsequent ligation-mediated PCR yielded short integration site fragments termed Int-tags, which were concatemerized for determining multiple integration sites in a single sequencing reaction. Analysis of chromosomal features and sequence preference associated with integration events confirmed the validity of the new high-throughput assay. The assay will aid the effort in understanding the mechanisms of target site selection during HIV-1 DNA integration, and the described methodology can be adapted easily to integration site studies involving other retroviruses and transposons.
Figures
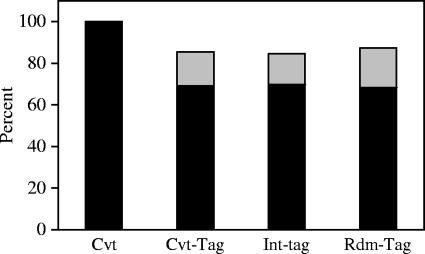
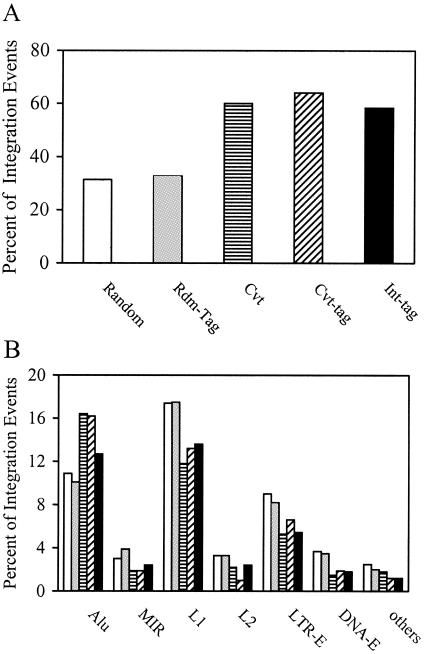
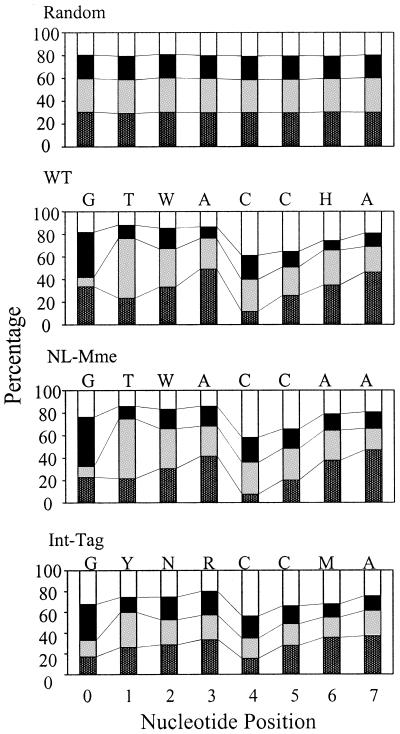
References
-
- Ausubel, F. A., R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman, J. A. Smith, and K. Struhl. 1999. Current protocols in molecular biology. Wiley, New York, N.Y.
-
- Brown, P. O. 1997. Integration, p. 161-204. In J. M. Coffin, S. H. Hughes, and H. E. Varmus (ed.), Retroviruses. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
-
- Bushman, F., M. Lewinski, A. Ciuffi, S. Barr, J. Leipzig, S. Hannenhalli, and C. Hoffmann. 2005. Genome-wide analysis of retroviral DNA integration. Nat. Rev. Microbiol. 3:848-858. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
