

Structural requirements for gp80 independence of human herpesvirus 8 interleukin-6 (vIL-6) and evidence for gp80 stabilization of gp130 signaling complexes induced by vIL-6
- PMID: 16973585
- PMCID: PMC1617266
- DOI: 10.1128/JVI.00872-06
Structural requirements for gp80 independence of human herpesvirus 8 interleukin-6 (vIL-6) and evidence for gp80 stabilization of gp130 signaling complexes induced by vIL-6
Abstract
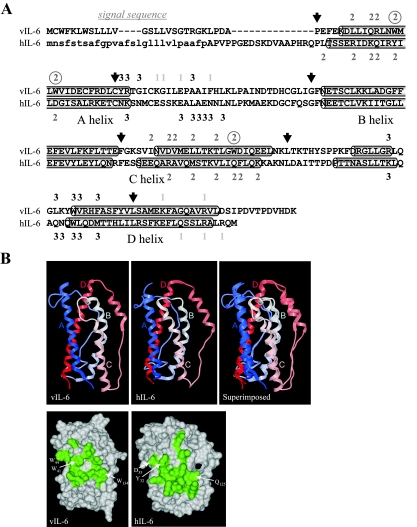
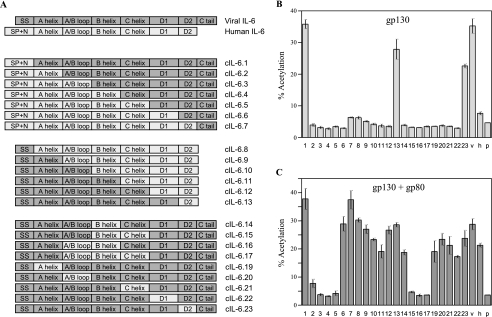
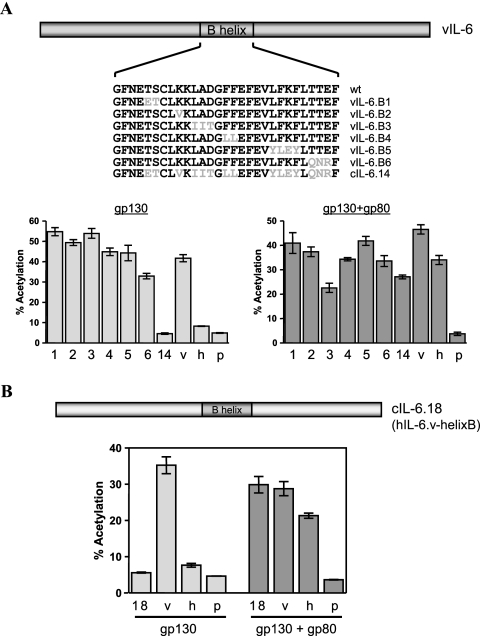
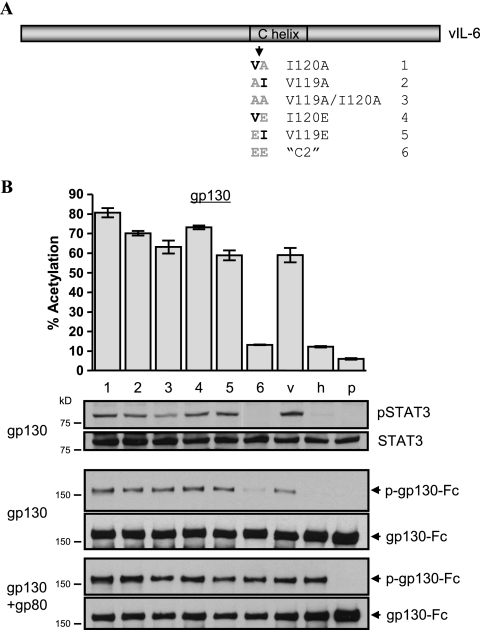
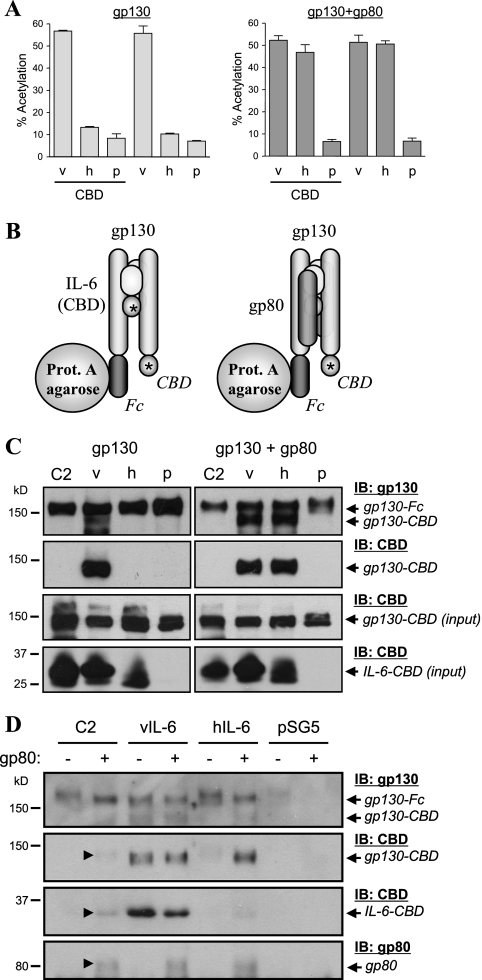
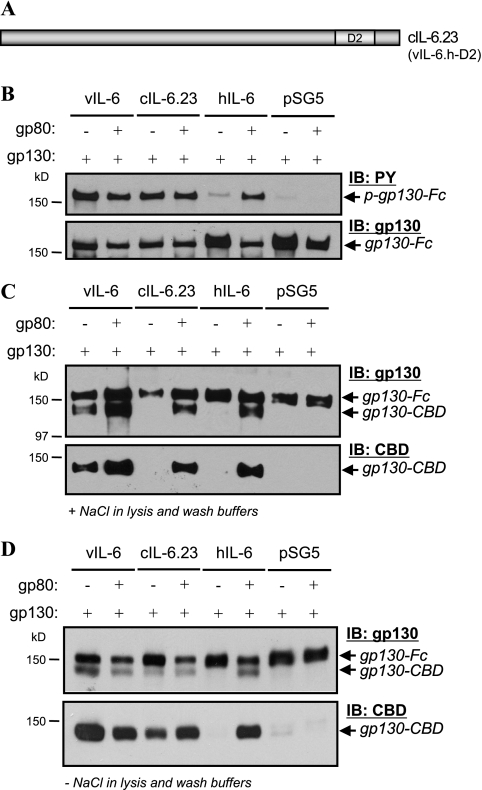
Human herpesvirus 8 interleukin-6 (vIL-6) displays 25% amino acid identity with human IL-6 (hIL-6) and shares an overall four-helix-bundle structure and gp130-mediated STAT/mitogen-activated protein kinase signaling with its cellular counterpart. However, vIL-6 is distinct in that it can signal through gp130 alone, in the absence of the nonsignaling gp80 alpha-subunit of the IL-6 receptor. To investigate the structural requirements for gp80 independence of vIL-6, a series of expression vectors encoding vIL-6/hIL-6 chimeric and site-mutated IL-6 proteins was generated. The replacement of hIL-6 residues with three vIL-6-specific tryptophans implicated in gp80 independence from crystallographic studies or the A and C helices containing these residues did not confer gp80 independence to hIL-6. The N- and C-terminal regions of vIL-6 could be substituted with hIL-6 sequences with the retention of gp80-independent signaling, but substitutions of other regions of vIL-6 (helix A, A/B loop, helix B, helix C, and proximal half of helix D) with equivalent sequences of hIL-6 abolished gp80 independence. Interestingly, the B helix of vIL-6 was absolutely required for gp80 independence, despite the fact that this region contains no receptor-binding residues. Point mutational analysis of helix C, which contains residues involved in physical and functional interactions with gp130 domains 2 and 3 (cytokine-binding homology region), identified a variant, VI120EE, that was able to signal and dimerize gp130 only in the presence of gp80. gp80 was also found to stabilize gp130:g130 dimers induced by a distal D helix variant of vIL-6 that was nonetheless able to signal independently of gp80. Together, our data reveal the crucial importance of overall vIL-6 structure and conformation for gp80-independent signaling and provide functional and physical evidence of the stabilization of vIL-6-induced gp130 signaling complexes by gp80.
Figures


References
-
- Aoki, Y., E. S. Jaffe, Y. Chang, K. Jones, J. Teruya-Feldstein, P. S. Moore, and G. Tosato. 1999. Angiogenesis and hematopoiesis induced by Kaposi's sarcoma-associated herpesvirus-encoded interleukin-6. Blood 93:4034-4043. - PubMed
-
- Aoki, Y., M. Narazaki, T. Kishimoto, and G. Tosato. 2001. Receptor engagement by viral interleukin-6 encoded by Kaposi's sarcoma-associated herpesvirus. Blood 98:3042-3049. - PubMed
-
- Aoki, Y., R. Yarchoan, K. Wyvill, S. Okamoto, R. F. Little, and G. Tosato. 2001. Detection of viral interleukin-6 in Kaposi's sarcoma-associated herpesvirus-linked disorders. Blood 97:2173-2176. - PubMed
-
- Boulanger, M. J., D. C. Chow, E. E., Brevnova, and K. C. Garcia. 2003. Hexameric structure and assembly of the interleukin-6/IL-6α-receptor/gp130 complex. Science 300:2101-2104. - PubMed
-
- Boulanger, M. J., D. C. Chow, E. Brevnova, M. Martick, G. Sandford, J. Nicholas, and K. C. Garcia. 2004. Molecular mechanisms for viral mimicry of a human cytokine: activation of gp130 by HHV-8 interleukin-6. J. Mol. Biol. 335:641-654. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
