

A full-genome phylogenetic analysis of varicella-zoster virus reveals a novel origin of replication-based genotyping scheme and evidence of recombination between major circulating clades
- PMID: 16973589
- PMCID: PMC1617253
- DOI: 10.1128/JVI.00715-06
A full-genome phylogenetic analysis of varicella-zoster virus reveals a novel origin of replication-based genotyping scheme and evidence of recombination between major circulating clades
Abstract
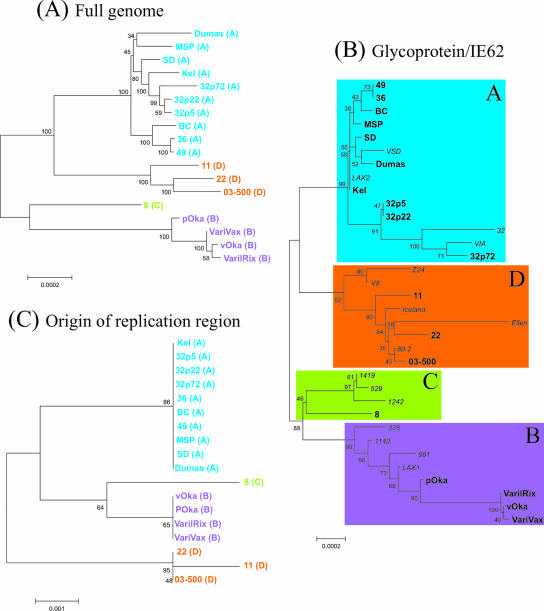
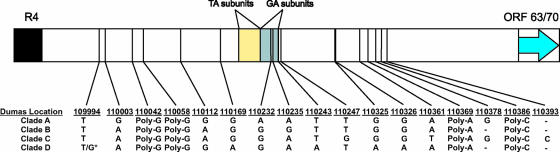
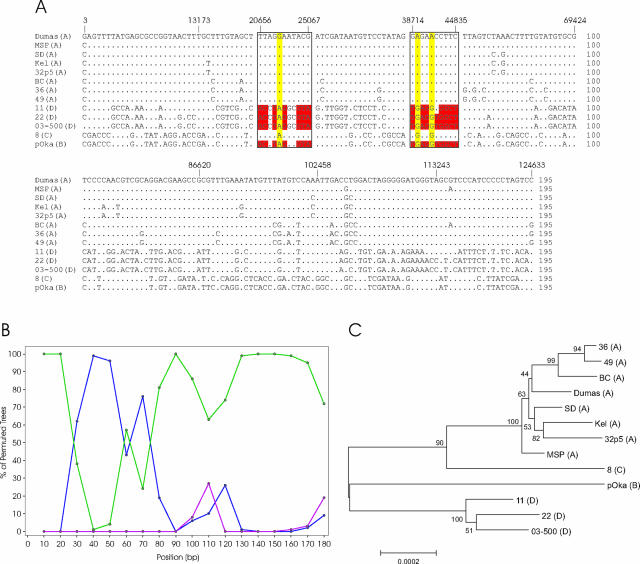
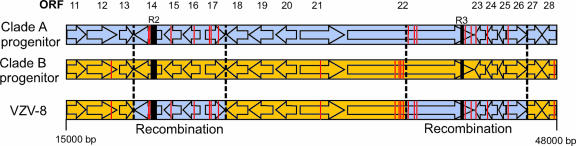
Varicella-zoster virus (VZV) is a remarkably stable virus that until recently was thought to exhibit near-universal genetic homogeneity among circulating wild-type strains. In recent years, the expanding knowledge of VZV genetics has led to a number of groups proposing sequence-based typing schemes, but no study has yet examined the relationships between VZV genotypes at a full-genome level. A central hypothesis of this study is that VZV has coevolved with humankind. In this study, 11 additional full VZV genomic sequences are presented, bringing the current number of complete genomic sequences publicly available to 18. The full-genome alignment contained strains representing four distinct clades, but the possibility exists that a fifth clade comprised of African and Asian-like isolates was not represented. A consolidated VZV genotyping scheme employing the origin-associated region between reiteration region R4 and open reading frames (ORFs) 63 and 70 is described, one which accurately categorizes strains into one of four clades related to the geographic origin of the isolates. The full-genome alignment also provided evidence for recombination having occurred between the major circulating VZV clades. One Canadian clinical isolate was primarily Asian-like in origin, with most of the genome showing strong sequence identity to the Japanese-like clade B, with the exceptions being two putative recombination regions, located in ORFs 14 to 17 and ORFs 22 to 26, which showed clear similarity to the European/North American clade A. The very low rate of single-nucleotide polymorphisms scattered across the genome made full-genome sequencing the only definitive method for identifying specific VZV recombination events.
Figures
References
-
- Argaw, T., J. I. Cohen, M. Klutch, K. Lekstrom, T. Yoshikawa, Y. Asano, and P. R. Krause. 2000. Nucleotide sequences that distinguish Oka vaccine from parental Oka and other varicella-zoster virus isolates. J. Infect. Dis. 181:1153-1157. - PubMed
-
- Baker, R. O., and J. D. Hall. 1998. Impaired mismatch extension by a herpes simplex DNA polymerase mutant with an editing nuclease defect. J. Biol. Chem. 273:24075-24082. - PubMed
-
- Barrett-Muir, W., K. Hawrami, J. Clarke, and J. Breuer. 2001. Investigation of varicella-zoster virus variation by heteroduplex mobility assay. Arch. Virol. Suppl. 17:17-25. - PubMed
-
- Barrett-Muir, W., F. T. Scott, P. Aaby, J. John, P. Matondo, Q. L. Chaudhry, M. Siqueira, A. Poulsen, K. Yaminishi, and J. Breuer. 2003. Genetic variation of varicella-zoster virus: evidence for geographical separation of strains. J. Med. Virol. 70:S42-S47. - PubMed
-
- Bowden, R., H. Sakaoka, P. Donnelly, and R. Ward. 2004. High recombination rate in herpes simplex virus type 1 natural populations suggests significant co-infection. Infect. Genet. Evol. 4:115-123. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
