

Ultra-high resolution imaging by fluorescence photoactivation localization microscopy
- PMID: 16980368
- PMCID: PMC1635685
- DOI: 10.1529/biophysj.106.091116
Ultra-high resolution imaging by fluorescence photoactivation localization microscopy
Abstract
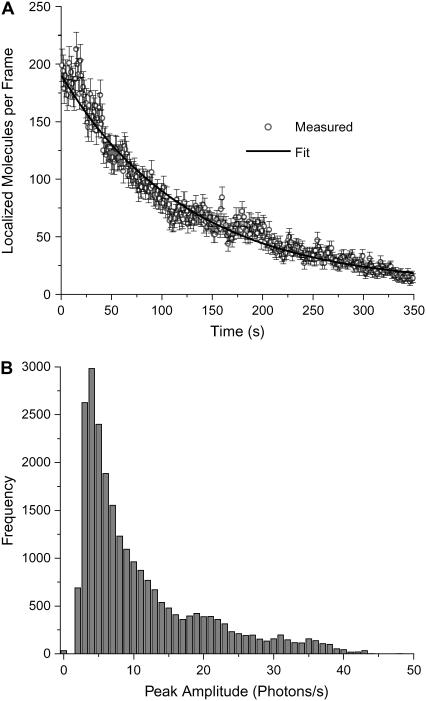
Biological structures span many orders of magnitude in size, but far-field visible light microscopy suffers from limited resolution. A new method for fluorescence imaging has been developed that can obtain spatial distributions of large numbers of fluorescent molecules on length scales shorter than the classical diffraction limit. Fluorescence photoactivation localization microscopy (FPALM) analyzes thousands of single fluorophores per acquisition, localizing small numbers of them at a time, at low excitation intensity. To control the number of visible fluorophores in the field of view and ensure that optically active molecules are separated by much more than the width of the point spread function, photoactivatable fluorescent molecules are used, in this case the photoactivatable green fluorescent protein (PA-GFP). For these photoactivatable molecules, the activation rate is controlled by the activation illumination intensity; nonfluorescent inactive molecules are activated by a high-frequency (405-nm) laser and are then fluorescent when excited at a lower frequency. The fluorescence is imaged by a CCD camera, and then the molecules are either reversibly inactivated or irreversibly photobleached to remove them from the field of view. The rate of photobleaching is controlled by the intensity of the laser used to excite the fluorescence, in this case an Ar+ ion laser. Because only a small number of molecules are visible at a given time, their positions can be determined precisely; with only approximately 100 detected photons per molecule, the localization precision can be as much as 10-fold better than the resolution, depending on background levels. Heterogeneities on length scales of the order of tens of nanometers are observed by FPALM of PA-GFP on glass. FPALM images are compared with images of the same molecules by widefield fluorescence. FPALM images of PA-GFP on a terraced sapphire crystal surface were compared with atomic force microscopy and show that the full width at half-maximum of features approximately 86 +/- 4 nm is significantly better than the expected diffraction-limited optical resolution. The number of fluorescent molecules and their brightness distribution have also been determined using FPALM. This new method suggests a means to address a significant number of biological questions that had previously been limited by microscope resolution.
Figures
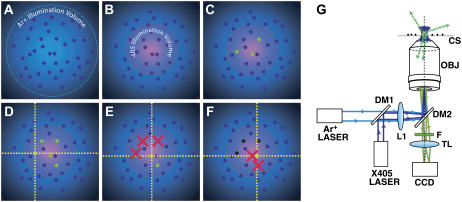
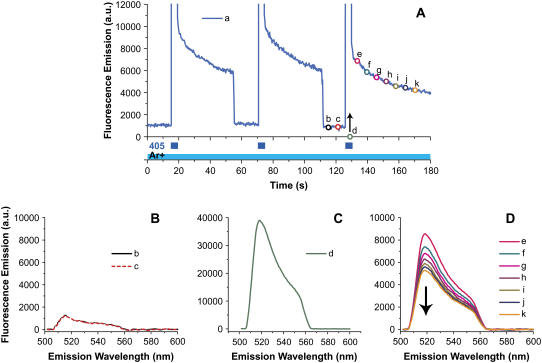
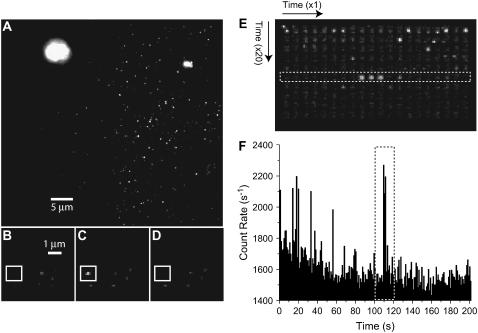
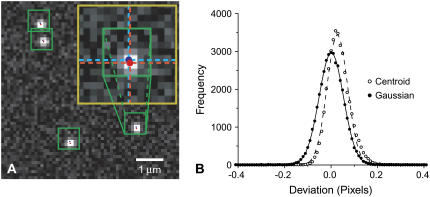
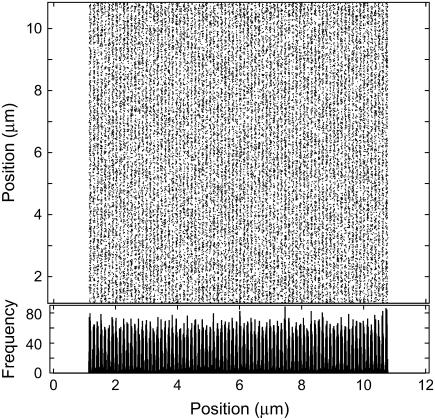
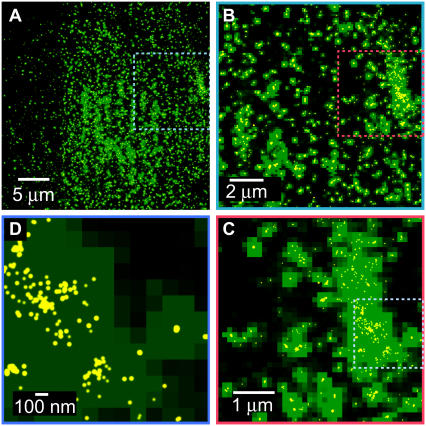
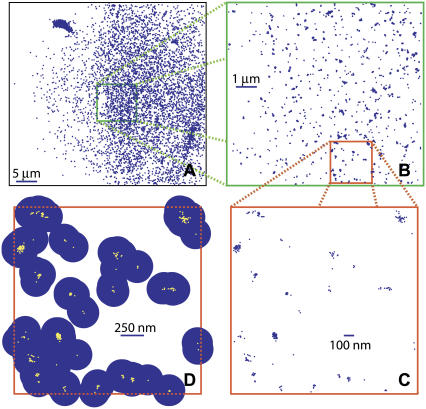
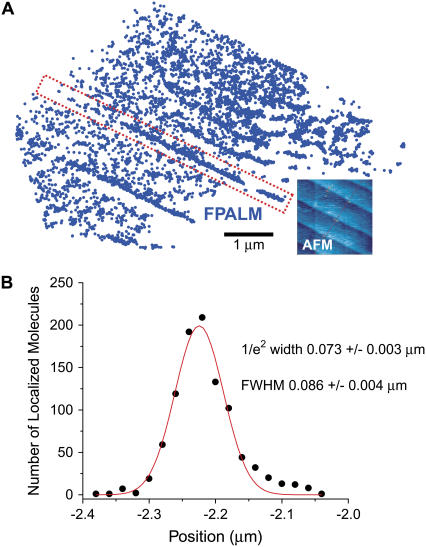
References
-
- Alberts, B. 2002. Molecular Biology of the Cell. Garland Science, New York.
-
- Richards, B., and E. Wolf. 1959. Electromagnetic diffraction in optical systems. 2. Structure of the image field in an aplanatic system. Proc. R. Soc. Lond. A Math. Phys. Sci. 253:358–379.
-
- Born, M., and E. Wolf. 1997. Principles of Optics: Electromagnetic Theory of Propagation, Interference and Diffraction of Light. Cambridge University Press, Cambridge, UK.
-
- Hell, S. W., M. Schrader, and H. T. van der Voort. 1997. Far-field fluorescence microscopy with three-dimensional resolution in the 100-nm range. J. Microsc. 187:1–7. - PubMed
-
- Schrader, M., and S. W. Hell. 1998. Three-dimensional super-resolution with a 4Pi-confocal microscope using image restoration. J. Appl. Phys. 84:4033–4042.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
