

Toward a more robust assessment of intraspecies diversity, using fewer genetic markers
- PMID: 16980418
- PMCID: PMC1636164
- DOI: 10.1128/AEM.01398-06
Toward a more robust assessment of intraspecies diversity, using fewer genetic markers
Abstract
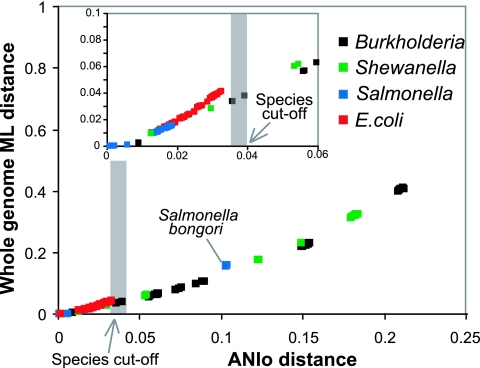
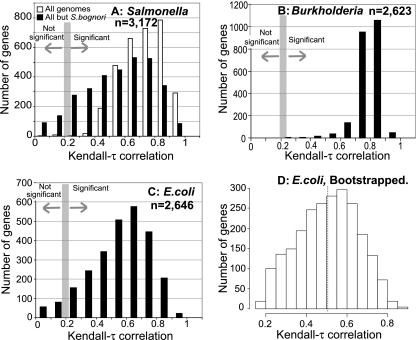
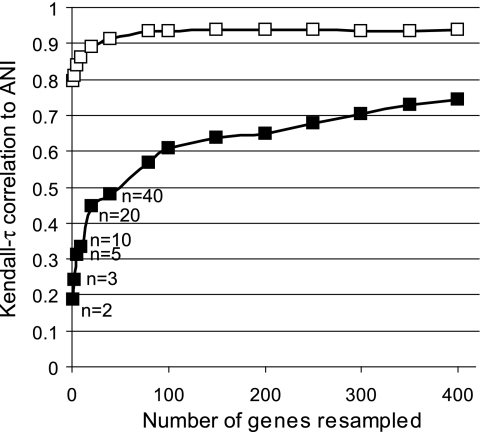
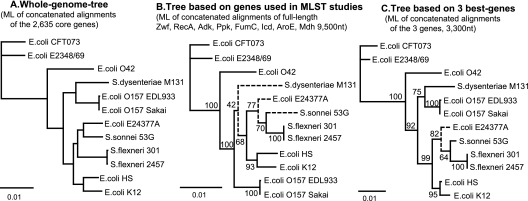
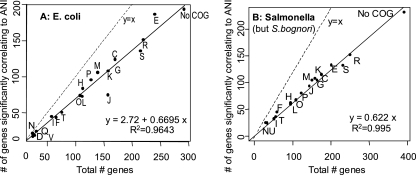
Phylogenetic sequence analysis of single or multiple genes has dominated the study and census of the genetic diversity among closely related bacteria. It remains unclear, however, how the results based on a few genes in the genome correlate with whole-genome-based relatedness and what genes (if any) best reflect whole-genome-level relatedness and hence should be preferentially used to economize on cost and to improve accuracy. We show here that phylogenies of closely related organisms based on the average nucleotide identity (ANI) of their shared genes correspond accurately to phylogenies based on state-of-the-art analysis of their whole-genome sequences. We use ANI to evaluate the phylogenetic robustness of every gene in the genome and show that almost all core genes, regardless of their functions and positions in the genome, offer robust phylogenetic reconstruction among strains that show 80 to 95% ANI (16S rRNA identity, >98.5%). Lack of elapsed time and, to a lesser extent, horizontal transfer and recombination make the selection of genes more critical for applications that target the intraspecies level, i.e., strains that show >95% ANI according to current standards. A much more accurate phylogeny for the Escherichia coli group was obtained based on just three best-performing genes according to our analysis compared to the concatenated alignment of eight genes that are commonly employed for phylogenetic purposes in this group. Our results are reproducible within the Salmonella, Burkholderia, and Shewanella groups and therefore are expected to have general applicability for microevolution studies, including metagenomic surveys.
Figures
References
-
- Adiri, R. S., U. Gophna, and E. Z. Ron. 2003. Multilocus sequence typing (MLST) of Escherichia coli O78 strains. FEMS Microbiol. Lett. 222:199-203. - PubMed
-
- Ciccarelli, F. D., T. Doerks, C. von Mering, C. J. Creevey, B. Snel, and P. Bork. 2006. Toward automatic reconstruction of a highly resolved tree of life. Science 311:1283-1287. - PubMed
-
- Feil, E. J. 2004. Small change: keeping pace with microevolution. Nat. Rev. Microbiol. 2:483-495. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
