

Comparison of multiple Amber force fields and development of improved protein backbone parameters
- PMID: 16981200
- PMCID: PMC4805110
- DOI: 10.1002/prot.21123
Comparison of multiple Amber force fields and development of improved protein backbone parameters
Abstract
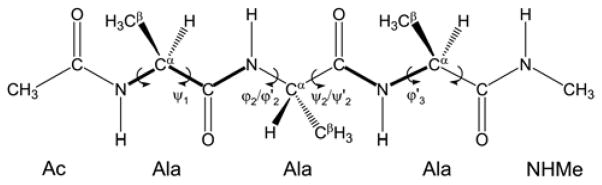
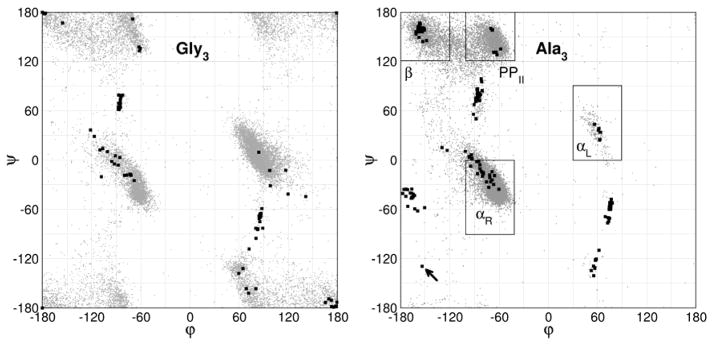
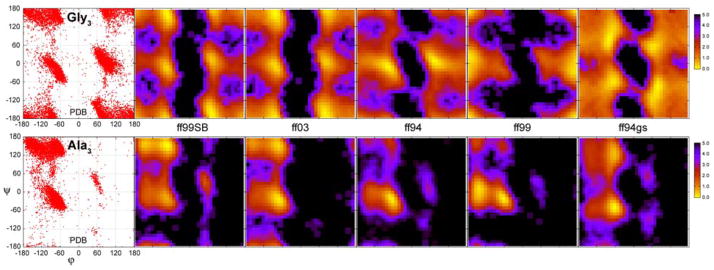
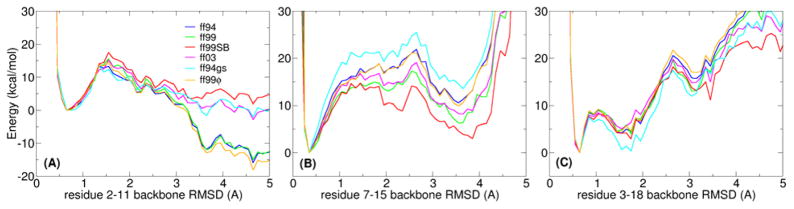
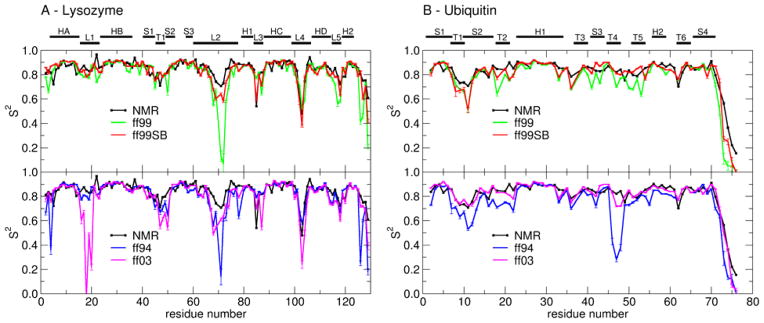
The ff94 force field that is commonly associated with the Amber simulation package is one of the most widely used parameter sets for biomolecular simulation. After a decade of extensive use and testing, limitations in this force field, such as over-stabilization of alpha-helices, were reported by us and other researchers. This led to a number of attempts to improve these parameters, resulting in a variety of "Amber" force fields and significant difficulty in determining which should be used for a particular application. We show that several of these continue to suffer from inadequate balance between different secondary structure elements. In addition, the approach used in most of these studies neglected to account for the existence in Amber of two sets of backbone phi/psi dihedral terms. This led to parameter sets that provide unreasonable conformational preferences for glycine. We report here an effort to improve the phi/psi dihedral terms in the ff99 energy function. Dihedral term parameters are based on fitting the energies of multiple conformations of glycine and alanine tetrapeptides from high level ab initio quantum mechanical calculations. The new parameters for backbone dihedrals replace those in the existing ff99 force field. This parameter set, which we denote ff99SB, achieves a better balance of secondary structure elements as judged by improved distribution of backbone dihedrals for glycine and alanine with respect to PDB survey data. It also accomplishes improved agreement with published experimental data for conformational preferences of short alanine peptides and better accord with experimental NMR relaxation data of test protein systems.
(c) 2006 Wiley-Liss, Inc.
Figures
References
-
- Rick SW, Stuart SJ. Potentials and algorithms for incorporating polarizability in computer simulations. Reviews in Computational Chemistry. 2002;18:89–146.
-
- Ponder JW, Case DA. Force fields for protein simulations. Protein Simulations. Volume 66. Advances in Protein Chemistry. 2003:27. - PubMed
-
- Mackerell AD. Empirical force fields for biological macromolecules: Overview and issues. Journal of Computational Chemistry. 2004;25(13):1584–1604. - PubMed
-
- Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. A Second Generation Force Field For the Simulation of Proteins, Nucleic Acids, and Organic Molecules. Journal of the American Chemical Society. 1995;117(19):5179–5197.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
