

Prostaglandin E2 induces fibroblast growth factor 9 via EP3-dependent protein kinase Cdelta and Elk-1 signaling
- PMID: 16982695
- PMCID: PMC1636777
- DOI: 10.1128/MCB.00941-06
Prostaglandin E2 induces fibroblast growth factor 9 via EP3-dependent protein kinase Cdelta and Elk-1 signaling
Abstract
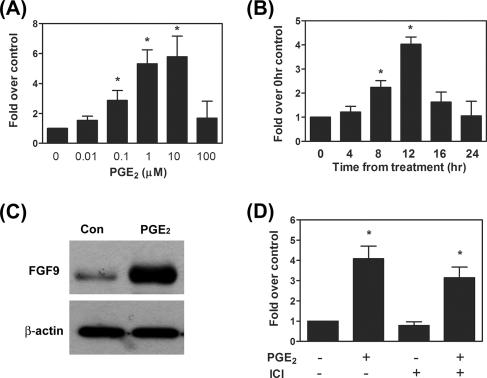
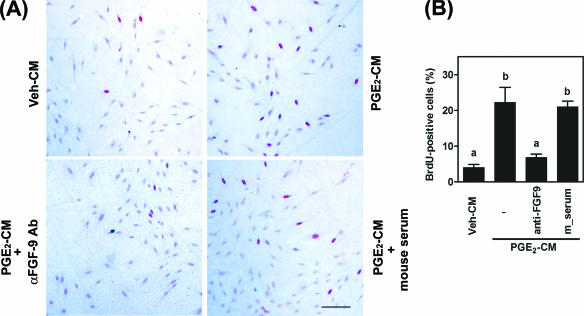
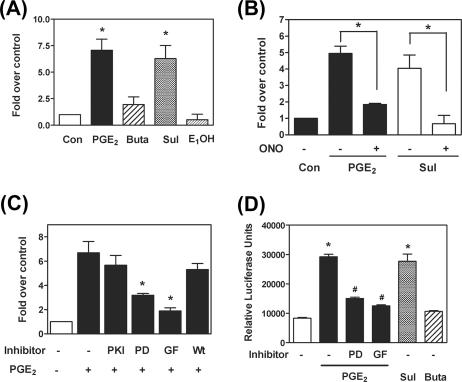
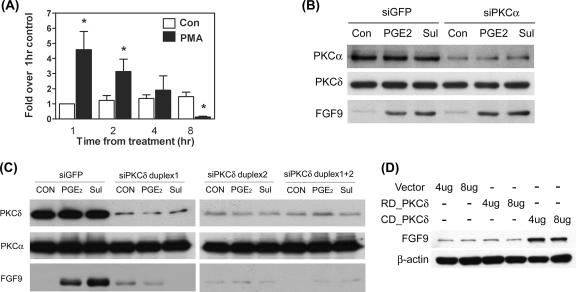
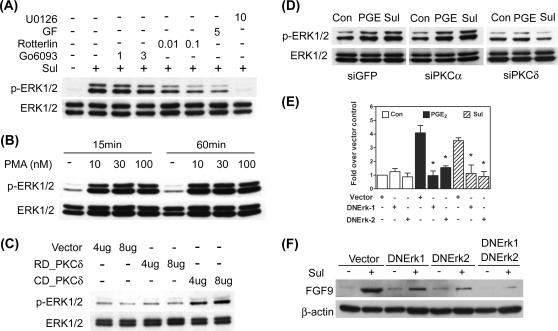
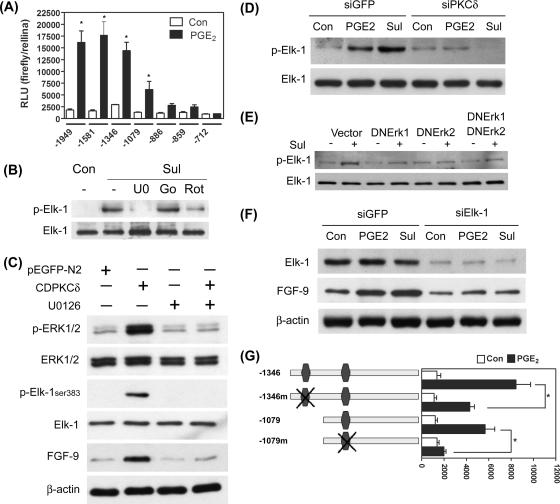
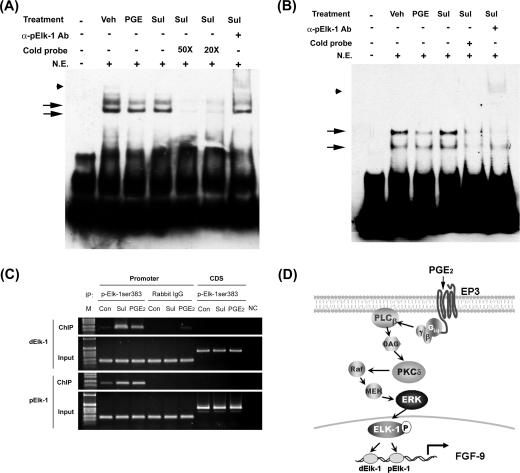
Fibroblast growth factor 9 (FGF-9) is a potent mitogen that controls the proper development of many tissues and organs. In contrast, aberrant expression of FGF-9 also results in the evolution of many human diseases, such as cancers and endometriosis. Despite its vital function being reported, the cellular and molecular mechanisms responsible for the regulation of FGF-9 expression are mostly unknown. We report here that prostaglandin E2 (PGE2) induces expression of FGF-9, which promotes endometriotic stromal cell proliferation, through the EP3 receptor-activated protein kinase Cdelta (PKCdelta) signaling pathway. Activation of PKCdelta leads to phosphorylation of ERK1/2, and the transcription factor Elk-1 thereby promotes transcription of FGF-9. Two Elk-1 cis-binding sites located at nucleotides -1324 to -1329 and -1046 to -1051 of the human FGF-9 promoter are identified as crucial for mediating PGE2 actions. Collectively, we demonstrate, for the first time, that PGE2 can directly induce FGF-9 expression via a novel signaling pathway involving EP3, PKCdelta, and a member of the ETS domain-containing transcription factor superfamily in primary human endometriotic stromal cells. Our findings may also provide a molecular framework for considering roles for PGE2 in FGF-9-related embryonic development and/or human diseases.
Figures
Similar articles
-
Protease-activated receptor-1 upregulates fibroblast growth factor 7 in stroma of benign prostatic hyperplasia.Prostate. 2008 Jul 1;68(10):1064-75. doi: 10.1002/pros.20767. Prostate. 2008. PMID: 18386288
-
Calcium influx into MIN6 insulinoma cells induces expression of Egr-1 involving extracellular signal-regulated protein kinase and the transcription factors Elk-1 and CREB.Eur J Cell Biol. 2009 Jan;88(1):19-33. doi: 10.1016/j.ejcb.2008.07.002. Epub 2008 Sep 9. Eur J Cell Biol. 2009. PMID: 18783846
-
Bradykinin induces matrix metalloproteinase-9 expression and cell migration through a PKC-delta-dependent ERK/Elk-1 pathway in astrocytes.Glia. 2008 Apr 15;56(6):619-32. doi: 10.1002/glia.20637. Glia. 2008. PMID: 18240315
-
Nuclear prostaglandin signaling system: biogenesis and actions via heptahelical receptors.Can J Physiol Pharmacol. 2003 Feb;81(2):196-204. doi: 10.1139/y02-163. Can J Physiol Pharmacol. 2003. PMID: 12710534 Review.
-
Structural interactomics: informatics approaches to aid the interpretation of genetic variation and the development of novel therapeutics.Mol Biosyst. 2009 Dec;5(12):1456-72. doi: 10.1039/B906402h. Epub 2009 Aug 6. Mol Biosyst. 2009. PMID: 19763326 Review.
Cited by
-
Inhibition of CD36-dependent phagocytosis by prostaglandin E2 contributes to the development of endometriosis.Am J Pathol. 2010 Feb;176(2):850-60. doi: 10.2353/ajpath.2010.090551. Epub 2009 Dec 24. Am J Pathol. 2010. PMID: 20035060 Free PMC article.
-
Overexpression of FGF9 in colon cancer cells is mediated by hypoxia-induced translational activation.Nucleic Acids Res. 2014 Mar;42(5):2932-44. doi: 10.1093/nar/gkt1286. Epub 2013 Dec 10. Nucleic Acids Res. 2014. PMID: 24334956 Free PMC article.
-
FGF-FGFR Mediates the Activity-Dependent Dendritogenesis of Layer IV Neurons during Barrel Formation.J Neurosci. 2017 Dec 13;37(50):12094-12105. doi: 10.1523/JNEUROSCI.1174-17.2017. Epub 2017 Nov 2. J Neurosci. 2017. PMID: 29097598 Free PMC article.
-
Maternal Recognition of Pregnancy in the Horse: Are MicroRNAs the Secret Messengers?Int J Mol Sci. 2020 Jan 9;21(2):419. doi: 10.3390/ijms21020419. Int J Mol Sci. 2020. PMID: 31936511 Free PMC article.
-
Induction of pyruvate dehydrogenase kinase-3 by hypoxia-inducible factor-1 promotes metabolic switch and drug resistance.J Biol Chem. 2008 Oct 17;283(42):28106-14. doi: 10.1074/jbc.M803508200. Epub 2008 Aug 21. J Biol Chem. 2008. PMID: 18718909 Free PMC article.
References
-
- Breyer, R. M., C. K. Bagdassarian, S. A. Myers, and M. D. Breyer. 2001. Prostanoid receptors: subtypes and signaling. Annu. Rev. Pharmacol. Toxicol. 41:661-690. - PubMed
-
- Castelao, J. E., R. D. Bart III, C. A. DiPerna, E. M. Sievers, and R. M. Bremner. 2003. Lung cancer and cyclooxygenase-2. Ann. Thorac. Surg. 76:1327-1335. - PubMed
-
- Castellone, M. D., H. Teramoto, B. O. Williams, K. M. Druey, and J. S. Gutkind. 2005. Prostaglandin E2 promotes colon cancer cell growth through a Gs-axin-beta-catenin signaling axis. Science 310:1504-1510. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous