

Expression of 4 genes between chromosome 15 breakpoints 1 and 2 and behavioral outcomes in Prader-Willi syndrome
- PMID: 16982806
- PMCID: PMC5453799
- DOI: 10.1542/peds.2006-0424
Expression of 4 genes between chromosome 15 breakpoints 1 and 2 and behavioral outcomes in Prader-Willi syndrome
Abstract
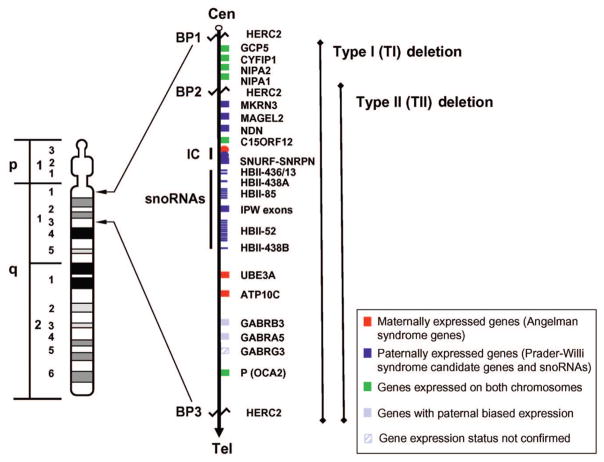
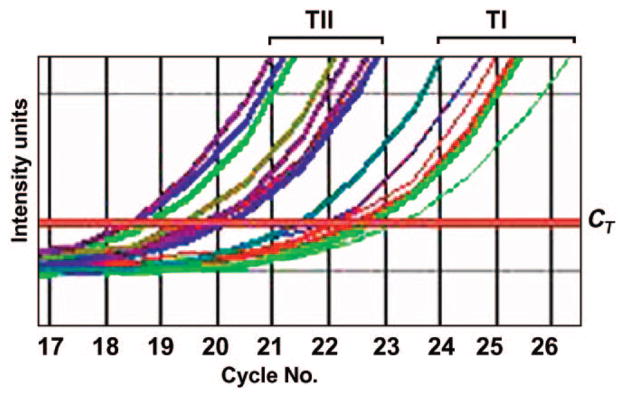
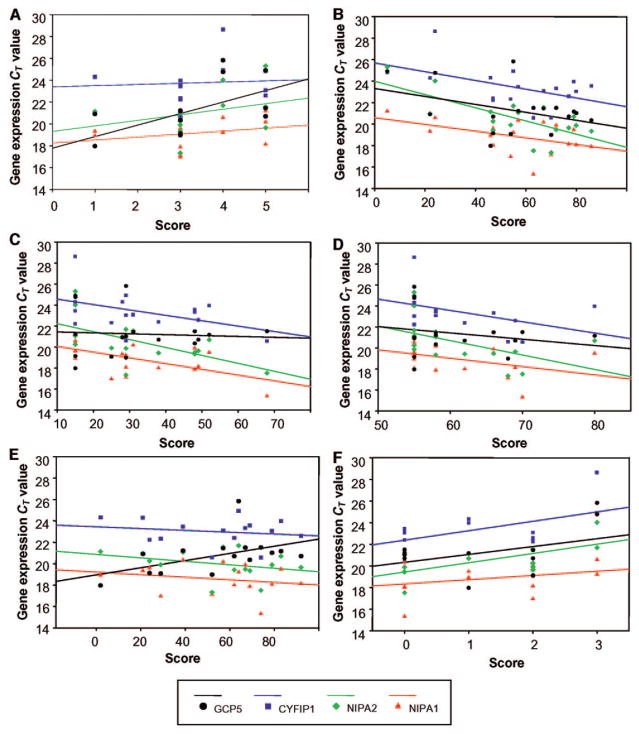
Prader-Willi syndrome is a neurodevelopmental disorder that is characterized by infantile hypotonia, feeding difficulties, hypogonadism, mental deficiency, hyperphagia (leading to obesity in early childhood), learning problems, and behavioral difficulties. A paternal 15q11-q13 deletion is found in approximately 70% of patients with Prader-Willi syndrome, approximately 25% have uniparental maternal disomy 15, and the remaining 2% to 5% have imprinting defects. The proximal deletion breakpoint in the 15q11-q13 region occurs at 1 of 2 sites located within either of 2 large duplicons allowing for the identification of 2 deletion subgroups. The larger, type I (TI) deletion involves breakpoint 1, which is close to the centromere, whereas the smaller, type II (TII) deletion involves breakpoint 2, located approximately 500 kilobases distal to breakpoint 1. Breakpoint 3 is located at the distal end of the 15q11-q13 region and common to both typical deletion subgroups. Analyses of the genetic subtypes of Prader-Willi syndrome to date have primarily compared individuals with typical deletion and uniparental maternal disomy 15 without grouping the individuals with a deletion into TI or TII. Distinct differences have been reported between individuals with Prader-Willi syndrome resulting from deletion compared with uniparental maternal disomy 15 in physical, cognitive, and behavioral parameters. We previously presented the first assessment of clinical differences in individuals with Prader-Willi syndrome categorized as having type I or II deletions. Adaptive behavior, obsessive-compulsive behaviors, reading, math, and visual-motor integration assessments were generally poorer in individuals with Prader-Willi syndrome and the TI deletion compared with subjects with Prader-Willi syndrome with the TII deletion or uniparental maternal disomy 15. Four genes (NIPA1, NIPA2, CYFIP1, and GCP5) have been identified in the chromosomal region between breakpoints 1 and 2 and are implicated in compulsive behavior and lower intellectual ability observed in individuals with Prader-Willi syndrome with TI versus TII deletions. We quantified messenger-RNA levels of these 4 genes in actively growing lymphoblastoid cells derived from 8 subjects with Prader-Willi syndrome with the TI deletion (4 males, 4 females; mean: age 25.2 +/- 8.9 years) and 9 with the TII deletion (3 males, 6 females; mean age: 19.5 +/- 5.8 years). Messenger-RNA levels were correlated with validated psychological and behavioral scales administered by trained psychologists blinded to genotype status. Messenger RNA from NIPA1, NIPA2, CYFIP1, and GCP5 was reduced but detectable in the subjects with Prader-Willi syndrome with the TI deletion, supporting biallelic expression. For the most part, messenger-RNA values were positively correlated with assessment parameters, indicating a direct relationship between messenger-RNA levels and better assessment scores, with the highest correlation for NIPA2. The coefficient of determination indicated the quantity of messenger RNA of the 4 genes explained from 24% to 99% of the variation of the behavioral and academic parameters measured. By comparison, the coefficient of determination for deletion type alone explained 5% to 50% of the variation in the assessed parameters. Understanding the influence of gene expression on behavioral and cognitive characteristics in humans is in the early stage of research development. Additional research is needed to identify the function of these genes and their interaction with gene networks to clarify the potential role they play in central nervous system development and function.
Figures
References
-
- Cassidy SB, Forsythe M, Heeger S, et al. Comparison of phenotype between patients with Prader-Willi syndrome due to deletion 15q and uniparental disomy 15. Am J Med Genet. 1997;68:433–440. - PubMed
-
- Nicholls RD, Knepper JL. Genome organization, function, and imprinting in Prader-Willi and Angelman syndromes. Annu Rev Genomics Hum Genet. 2001;2:153–175. - PubMed
-
- Chai JH, Locke DP, Greally JM, et al. Identification of four highly conserved genes between breakpoint hotspots BP1 and BP2 of the Prader-Willi/Angelman syndromes deletion region that have undergone evolutionary transposition mediated by flanking duplicons. Am J Hum Genet. 2003;73:898–925. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
