

Coding limits on the number of transcription factors
- PMID: 16984633
- PMCID: PMC1590034
- DOI: 10.1186/1471-2164-7-239
Coding limits on the number of transcription factors
Abstract
Background: Transcription factor proteins bind specific DNA sequences to control the expression of genes. They contain DNA binding domains which belong to several super-families, each with a specific mechanism of DNA binding. The total number of transcription factors encoded in a genome increases with the number of genes in the genome. Here, we examined the number of transcription factors from each super-family in diverse organisms.
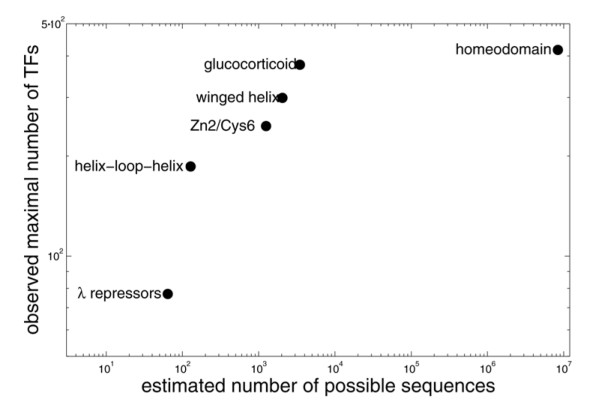
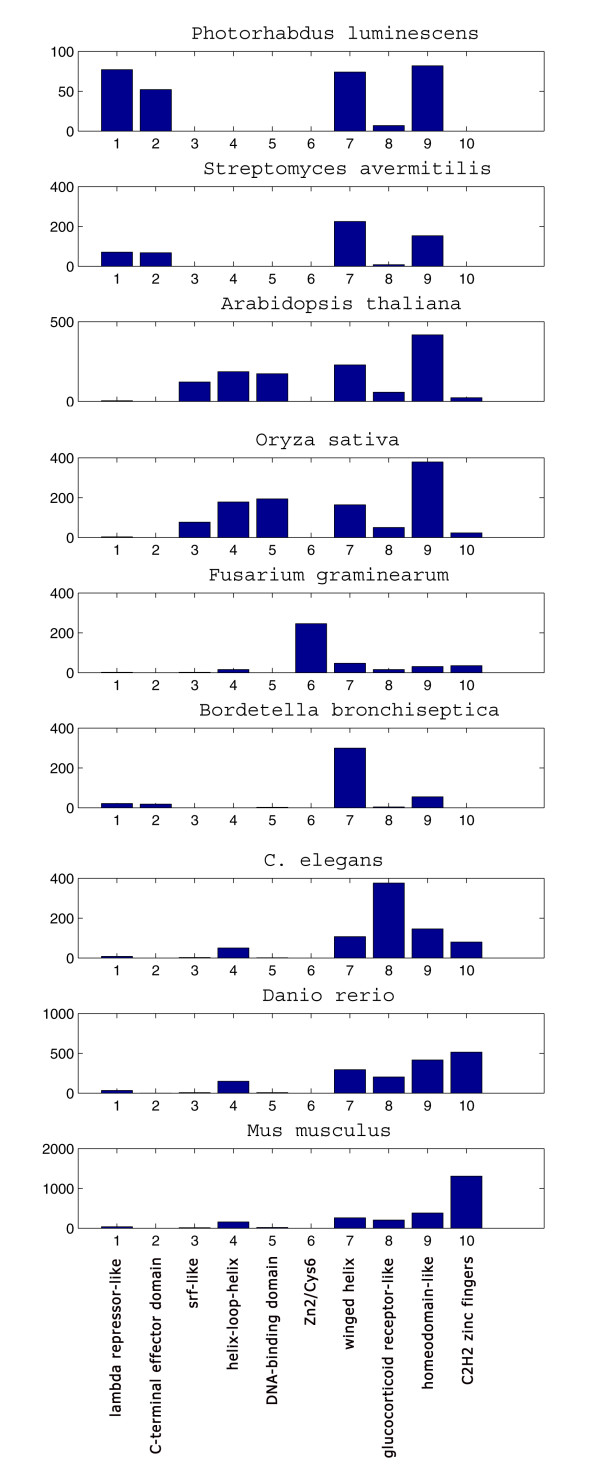
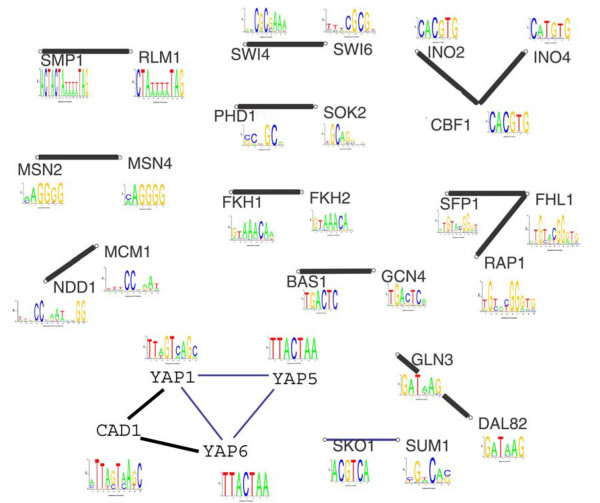
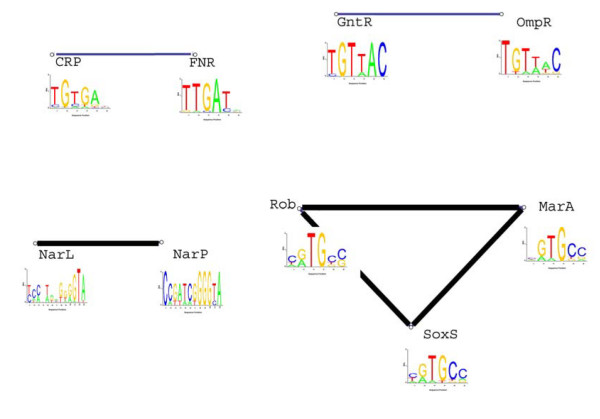
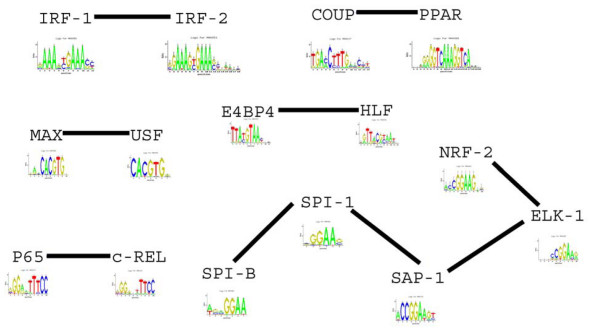
Results: We find that the number of transcription factors from most super-families appears to be bounded. For example, the number of winged helix factors does not generally exceed 300, even in very large genomes. The magnitude of the maximal number of transcription factors from each super-family seems to correlate with the number of DNA bases effectively recognized by the binding mechanism of that super-family. Coding theory predicts that such upper bounds on the number of transcription factors should exist, in order to minimize cross-binding errors between transcription factors. This theory further predicts that factors with similar binding sequences should tend to have similar biological effect, so that errors based on mis-recognition are minimal. We present evidence that transcription factors with similar binding sequences tend to regulate genes with similar biological functions, supporting this prediction.
Conclusion: The present study suggests limits on the transcription factor repertoire of cells, and suggests coding constraints that might apply more generally to the mapping between binding sites and biological function.
Figures

References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
