

Role of toll-like receptor signalling in Abeta uptake and clearance
- PMID: 16984903
- PMCID: PMC2445613
- DOI: 10.1093/brain/awl249
Role of toll-like receptor signalling in Abeta uptake and clearance
Abstract
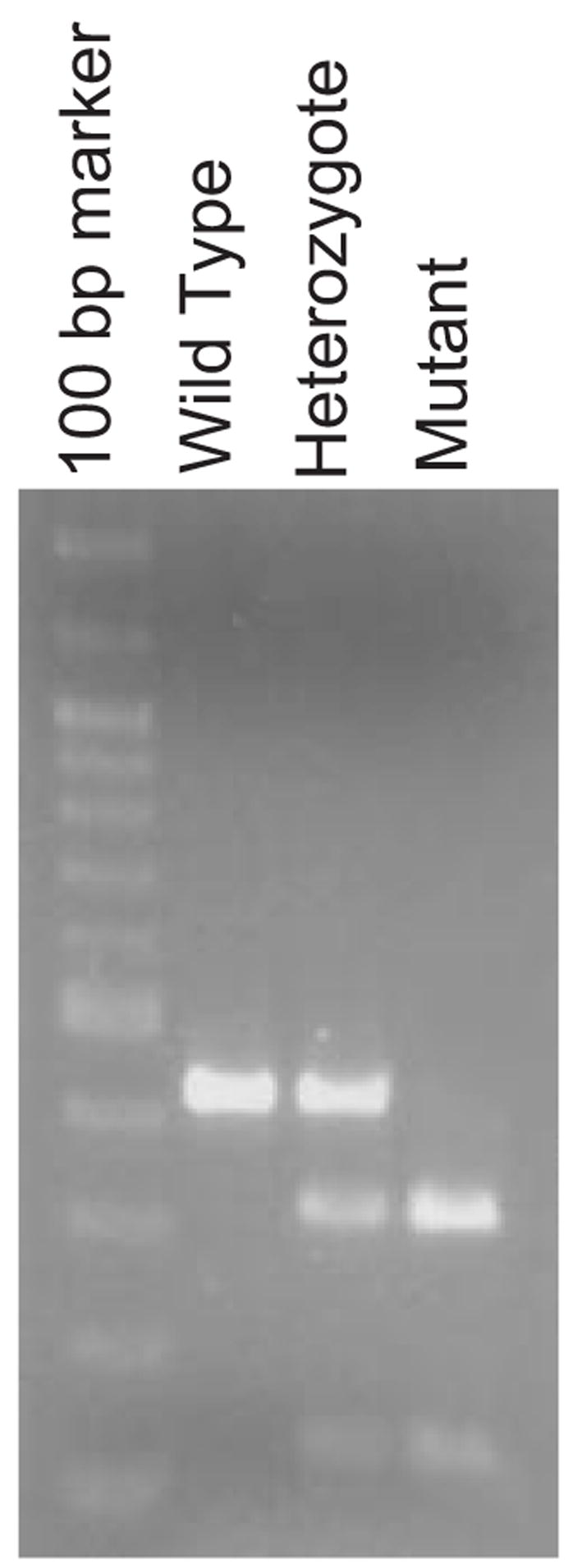
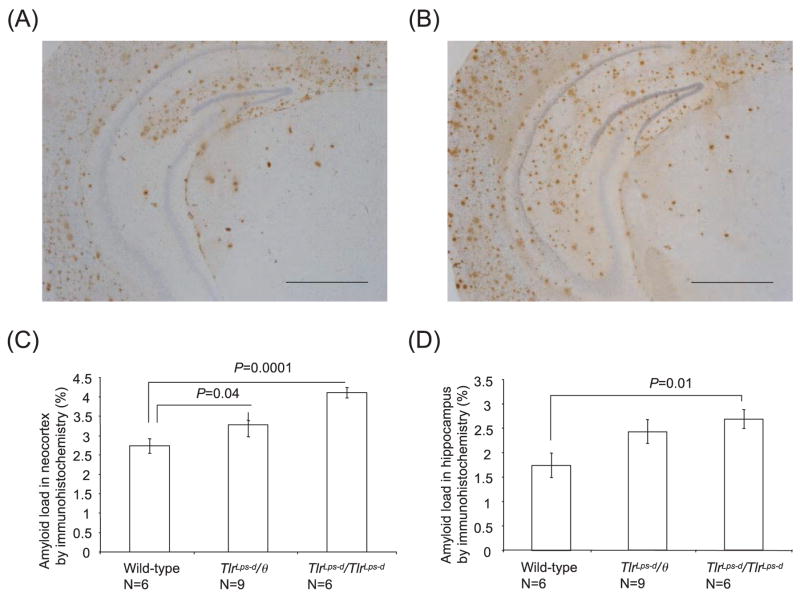
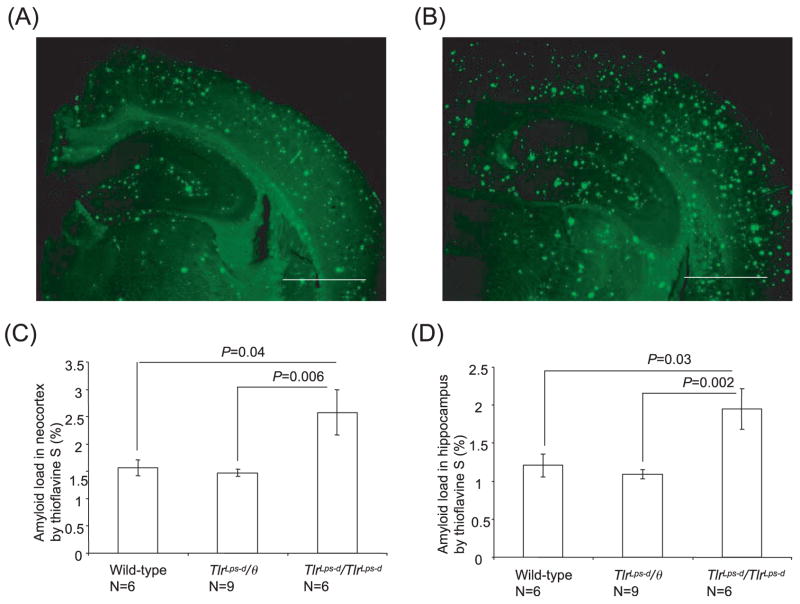
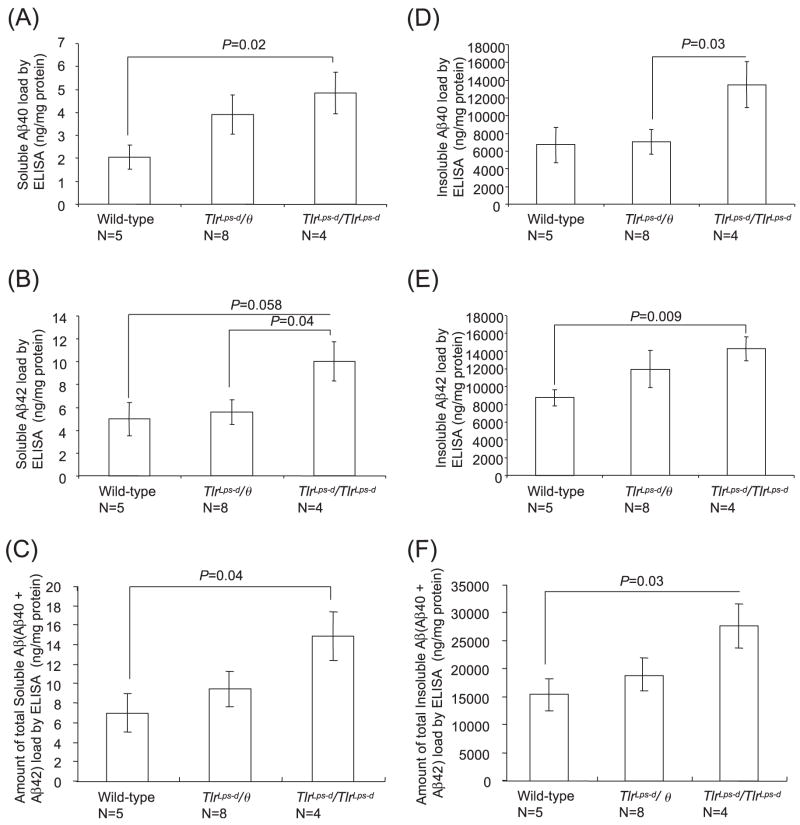
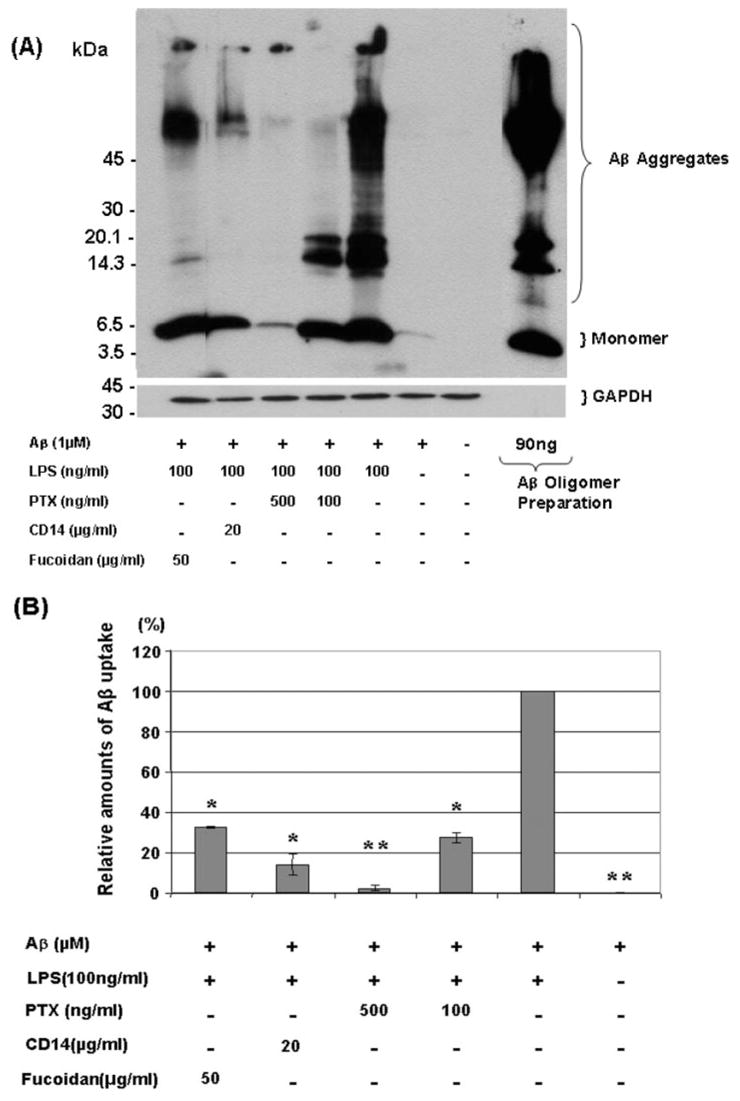
Deposits of amyloid beta-protein (Abeta) in neuritic plaques and cerebral vessels are a pathological hallmark of Alzheimer's disease. Fibrillar Abeta deposits are closely associated with inflammatory responses such as activated microglia in brain with this disease. Increasing lines of evidence support the hypothesis that activated microglia, innate immune cells in the CNS, play a pivotal role in the progression of the disease: either clearing Abeta deposits by phagocytic activity or releasing cytotoxic substances and pro-inflammatory cytokines. Toll-like receptors (TLRs) are a family of pattern-recognition receptors in the innate immune system. Exogenous and endogenous TLR ligands activate microglia. To investigate the role of TLR4 in the amyloidogenesis in vivo, we determined the amounts of cerebral Abeta in Alzheimer's disease mouse models with different genotypes of TLR4 using three distinct methods. We show that mouse models (Mo/Hu APPswe PS1dE9 mice) homozygous for a destructive mutation of TLR4 (Tlr(Lps-d)/Tlr(Lps-d)) had increases in diffuse and fibrillar Abeta deposits by immunocytochemistry, fibrillar Abeta deposits by thioflavine-S staining and buffer-soluble and insoluble Abeta by ELISA in the cerebrum, as compared with TLR4 wild-type mouse models. Although the differences in these parameters were less significant, mouse models heterozygous for the mutation (Tlr(Lps-d)/) showed co-dominant phenotypes. Consistent with these observations in vivo, cultured microglia derived from Tlr(Lps-d)/Tlr(Lps-d) mice failed to show an increase in Abeta uptake after stimulation with a TLR4 ligand but not with a TLR9 ligand in vitro. Furthermore, activation of microglia (BV-2 cell) with a TLR2, TLR4 or TLR9 ligand, markedly boosted ingestion of Abeta in vitro. These results suggest that TLR signalling pathway(s) may be involved in clearance of Abeta-deposits in the brain and that TLRs can be a therapeutic target for Alzheimer's disease.
Figures
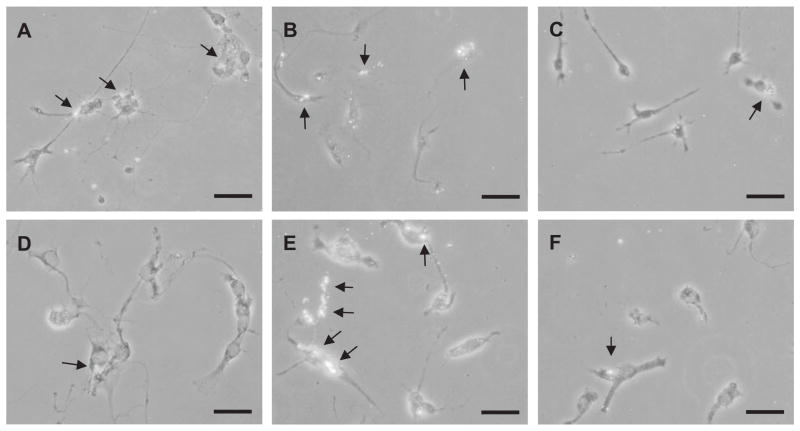
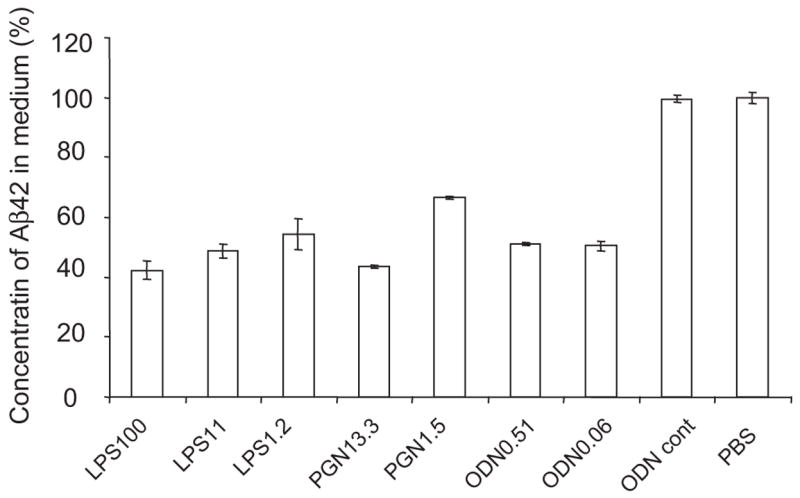
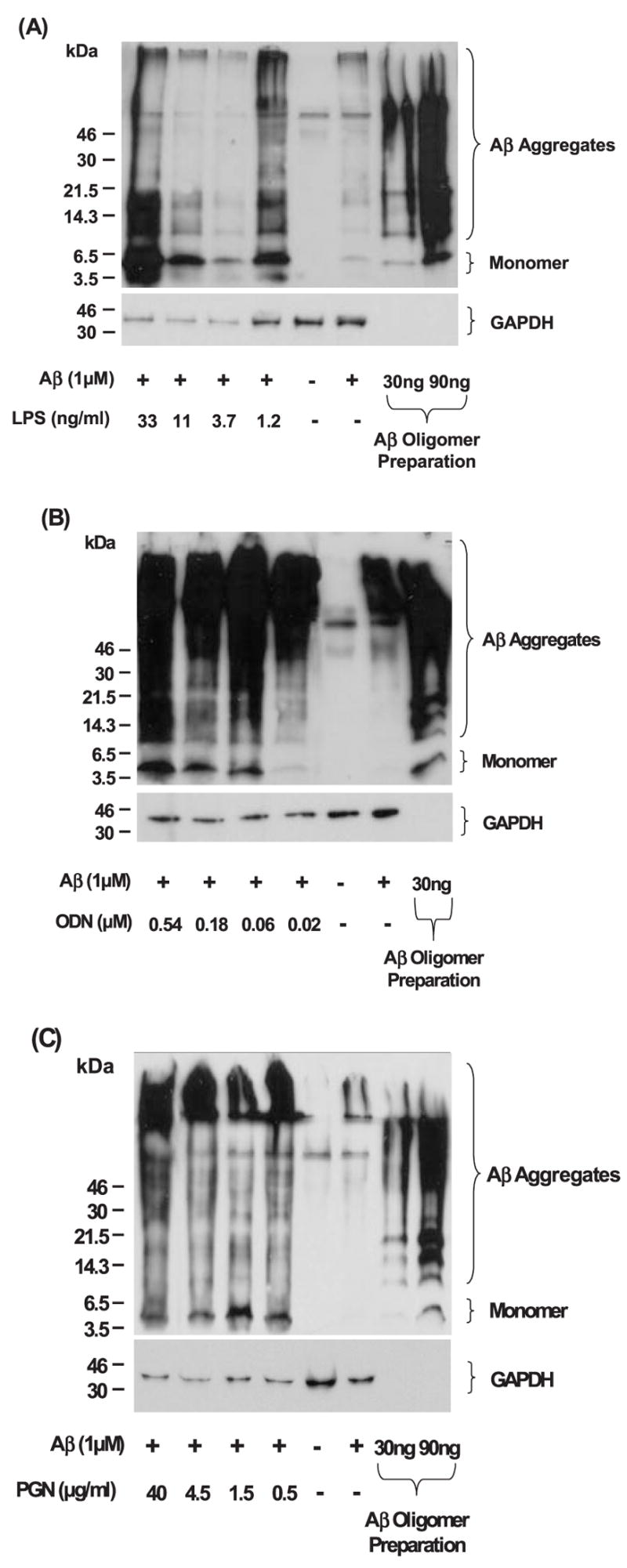
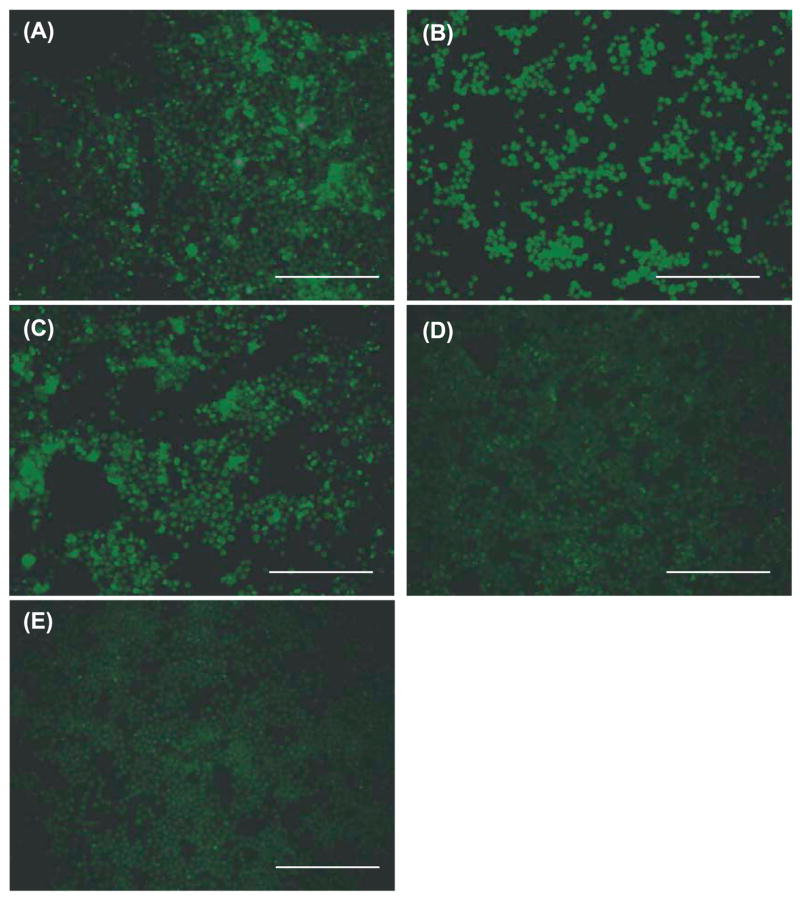
References
-
- Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. - PubMed
-
- Ard MD, Cole GM, Wei J, Mehrle AP, Fratkin JD. Scavenging of Alzheimer’s amyloid beta-protein by microglia in culture. J Neurosci Res. 1996;43:190–202. - PubMed
-
- Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, et al. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6:916–9. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
