

Using RNA secondary structures to guide sequence motif finding towards single-stranded regions
- PMID: 16987907
- PMCID: PMC1903381
- DOI: 10.1093/nar/gkl544
Using RNA secondary structures to guide sequence motif finding towards single-stranded regions
Abstract
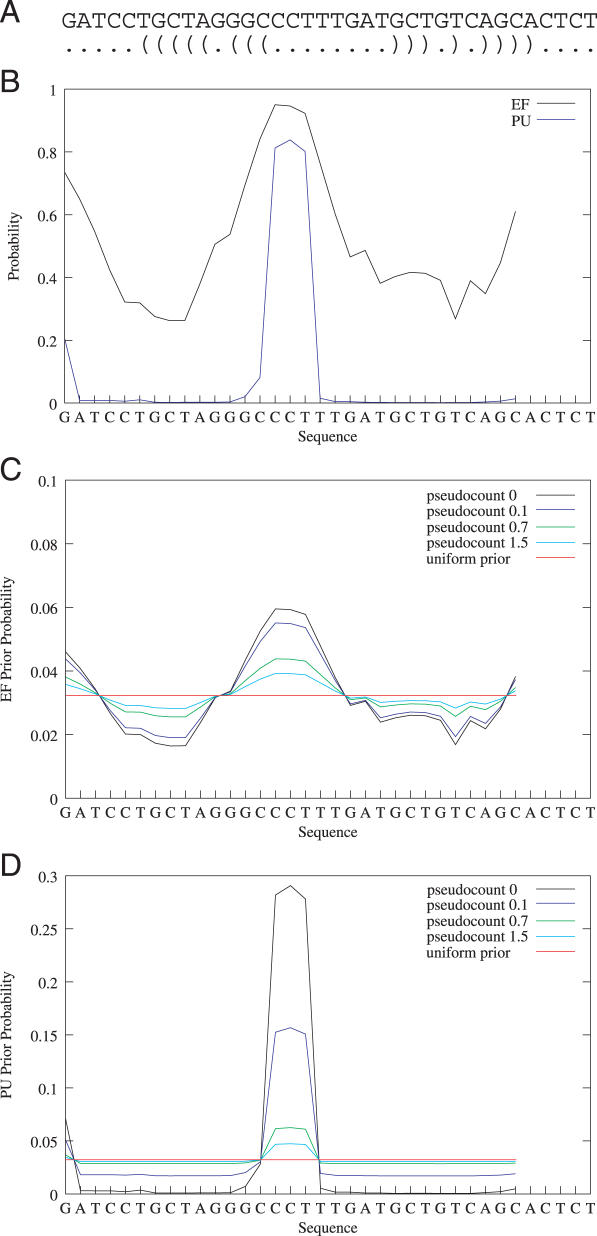
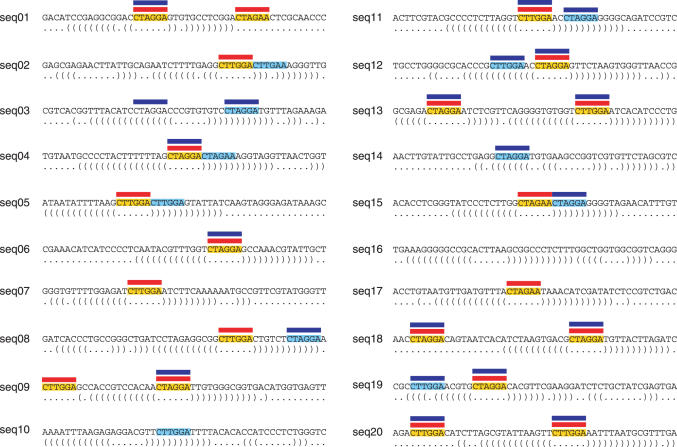
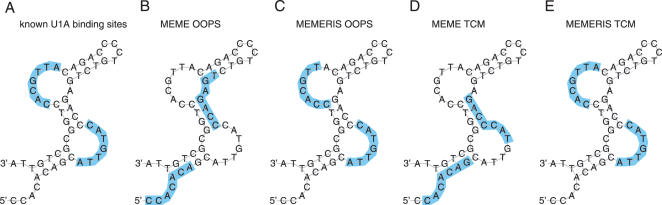
RNA binding proteins recognize RNA targets in a sequence specific manner. Apart from the sequence, the secondary structure context of the binding site also affects the binding affinity. Binding sites are often located in single-stranded RNA regions and it was shown that the sequestration of a binding motif in a double-strand abolishes protein binding. Thus, it is desirable to include knowledge about RNA secondary structures when searching for the binding motif of a protein. We present the approach MEMERIS for searching sequence motifs in a set of RNA sequences and simultaneously integrating information about secondary structures. To abstract from specific structural elements, we precompute position-specific values measuring the single-strandedness of all substrings of an RNA sequence. These values are used as prior knowledge about the motif starts to guide the motif search. Extensive tests with artificial and biological data demonstrate that MEMERIS is able to identify motifs in single-stranded regions even if a stronger motif located in double-strand parts exists. The discovered motif occurrences in biological datasets mostly coincide with known protein-binding sites. This algorithm can be used for finding the binding motif of single-stranded RNA-binding proteins in SELEX or other biological sequence data.
Figures


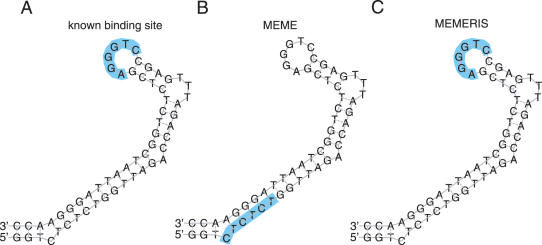
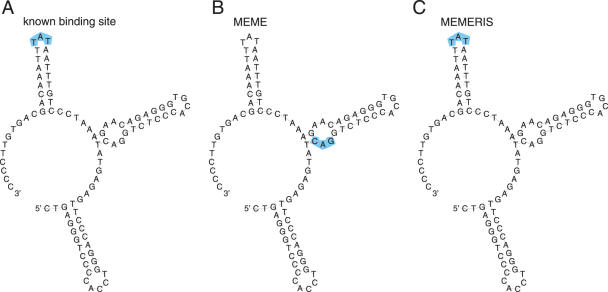
References
-
- Messias A.C., Sattler M. Structural basis of single-stranded RNA recognition. Acc. Chem. Res. 2004;37:279–287. - PubMed
-
- Hall K.B. RNA-protein interactions. Curr. Opin. Struct. Biol. 2002;12:283–288. - PubMed
-
- Thisted T., Lyakhov D.L., Liebhaber S.A. Optimized RNA targets of two closely related triple KH domain proteins, heterogeneous nuclear ribonucleoprotein K and alphaCP-2KL, suggest distinct modes of RNA recognition. J. Biol. Chem. 2001;276:17484–17496. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
