

A novel automated lazy learning QSAR (ALL-QSAR) approach: method development, applications, and virtual screening of chemical databases using validated ALL-QSAR models
- PMID: 16995729
- PMCID: PMC2536695
- DOI: 10.1021/ci060132x
A novel automated lazy learning QSAR (ALL-QSAR) approach: method development, applications, and virtual screening of chemical databases using validated ALL-QSAR models
Abstract
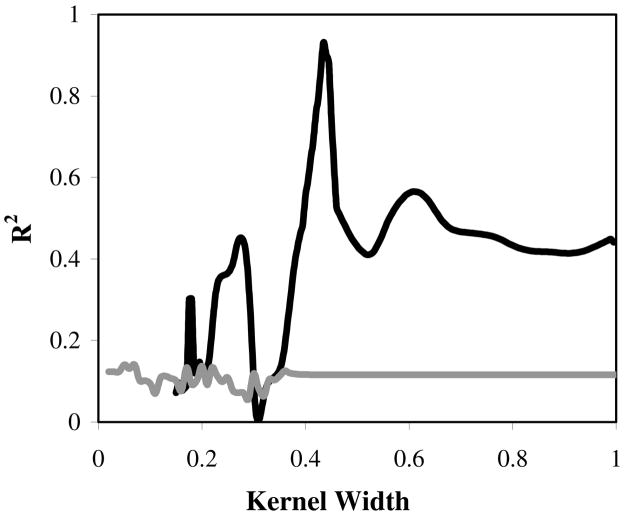
A novel automated lazy learning quantitative structure-activity relationship (ALL-QSAR) modeling approach has been developed on the basis of the lazy learning theory. The activity of a test compound is predicted from a locally weighted linear regression model using chemical descriptors and the biological activity of the training set compounds most chemically similar to this test compound. The weights with which training set compounds are included in the regression depend on the similarity of those compounds to a test compound. We have applied the ALL-QSAR method to several experimental chemical data sets including 48 anticonvulsant agents with known ED50 values, 48 dopamine D1-receptor antagonists with known competitive binding affinities (Ki), and a Tetrahymena pyriformis data set containing 250 phenolic compounds with toxicity IGC50 values. When applied to database screening, models developed for anticonvulsant agents identified several known anticonvulsant compounds that were not only absent in the training set but highly chemically dissimilar to the training set compounds. This initial success indicates that ALL-QSAR can be further exploited as a general tool for accurate bioactivity prediction and database screening in drug design and discovery. Because of its local nature, the ALL-QSAR approach appears to be especially well-suited for the development of highly predictive models for the sparse or unevenly distributed data sets.
Figures













Similar articles
-
Combinatorial QSAR modeling of specificity and subtype selectivity of ligands binding to serotonin receptors 5HT1E and 5HT1F.J Chem Inf Model. 2008 May;48(5):997-1013. doi: 10.1021/ci700404c. Epub 2008 May 10. J Chem Inf Model. 2008. PMID: 18470978
-
Application of predictive QSAR models to database mining: identification and experimental validation of novel anticonvulsant compounds.J Med Chem. 2004 Apr 22;47(9):2356-64. doi: 10.1021/jm030584q. J Med Chem. 2004. PMID: 15084134
-
Combinatorial QSAR modeling of chemical toxicants tested against Tetrahymena pyriformis.J Chem Inf Model. 2008 Apr;48(4):766-84. doi: 10.1021/ci700443v. Epub 2008 Mar 1. J Chem Inf Model. 2008. PMID: 18311912
-
Predictive QSAR modeling workflow, model applicability domains, and virtual screening.Curr Pharm Des. 2007;13(34):3494-504. doi: 10.2174/138161207782794257. Curr Pharm Des. 2007. PMID: 18220786 Review.
-
QSAR and 3D-QSAR studies applied to compounds with anticonvulsant activity.Expert Opin Drug Discov. 2015 Jan;10(1):37-51. doi: 10.1517/17460441.2015.968123. Epub 2014 Oct 9. Expert Opin Drug Discov. 2015. PMID: 25297377 Review.
Cited by
-
Predicting Cytotoxicity of Metal Oxide Nanoparticles using Isalos Analytics Platform.Nanomaterials (Basel). 2020 Oct 13;10(10):2017. doi: 10.3390/nano10102017. Nanomaterials (Basel). 2020. PMID: 33066094 Free PMC article.
-
QSAR study of anti-Human African Trypanosomiasis activity for 2-phenylimidazopyridines derivatives using DFT and Lipinski's descriptors.Heliyon. 2019 Mar 7;5(3):e01304. doi: 10.1016/j.heliyon.2019.e01304. eCollection 2019 Mar. Heliyon. 2019. PMID: 30899832 Free PMC article.
-
Assessing the Effects of Alloxydim Phototransformation Products by QSAR Models and a Phytotoxicity Study.Molecules. 2018 Apr 24;23(5):993. doi: 10.3390/molecules23050993. Molecules. 2018. PMID: 29695081 Free PMC article.
-
Computational modeling of novel inhibitors targeting the Akt pleckstrin homology domain.Bioorg Med Chem. 2009 Oct 1;17(19):6983-92. doi: 10.1016/j.bmc.2009.08.022. Epub 2009 Aug 19. Bioorg Med Chem. 2009. PMID: 19734051 Free PMC article.
-
Machine learning prediction of intestinal α-glucosidase inhibitors using a diverse set of ligands: a drug repurposing effort with drugBank database screening.In Silico Pharmacol. 2025 Jun 25;13(2):95. doi: 10.1007/s40203-025-00384-8. eCollection 2025. In Silico Pharmacol. 2025. PMID: 40575395 Free PMC article.
References
-
- Dietrich SW, Dreyer ND, Hansch C, Bentley DL. Confidence-Interval Estimators for Parameters Associated with Quantitative Structure-Activity-Relationships. J Med Chem. 1980;23:1201–1205. - PubMed
-
- Hadjipavloulitina D, Hansch C. Quantitative Structure-Activity-Relationships of the Benzodiazepines - A Review and Reevaluation. Chem Rev. 1994;94:1483–1505.
-
- Hansch C, Muir RM, Fujita T, Maloney PP, Geiger E, Streich M. The Correlation of Biological Activity of Plant Growth Regulators and Chloromycetin Derivatives with Hammett Constants and Partition Coefficients. J Am Chem Soc. 1963;85:2817–2824.
-
- Hansch C, Kurup A, Garg R, Gao H. Chem-bioinformatics and QSAR: A review of QSAR lacking positive hydrophobic terms. Chem Rev. 2001;101:619–672. - PubMed
-
- Hansch C, Leo A, Mekapati SB, Kurup A. Qsar and Adme. Bioorg Med Chem. 2004;12:3391–3400. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
