

Sampling realistic protein conformations using local structural bias
- PMID: 17002495
- PMCID: PMC1570370
- DOI: 10.1371/journal.pcbi.0020131
Sampling realistic protein conformations using local structural bias
Abstract
The prediction of protein structure from sequence remains a major unsolved problem in biology. The most successful protein structure prediction methods make use of a divide-and-conquer strategy to attack the problem: a conformational sampling method generates plausible candidate structures, which are subsequently accepted or rejected using an energy function. Conceptually, this often corresponds to separating local structural bias from the long-range interactions that stabilize the compact, native state. However, sampling protein conformations that are compatible with the local structural bias encoded in a given protein sequence is a long-standing open problem, especially in continuous space. We describe an elegant and mathematically rigorous method to do this, and show that it readily generates native-like protein conformations simply by enforcing compactness. Our results have far-reaching implications for protein structure prediction, determination, simulation, and design.
Conflict of interest statement
Figures
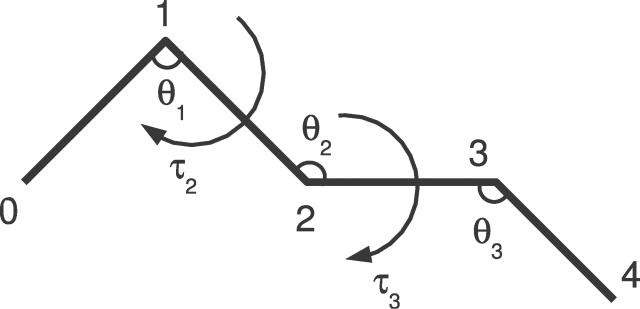
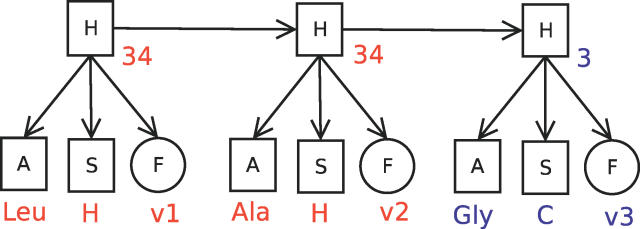
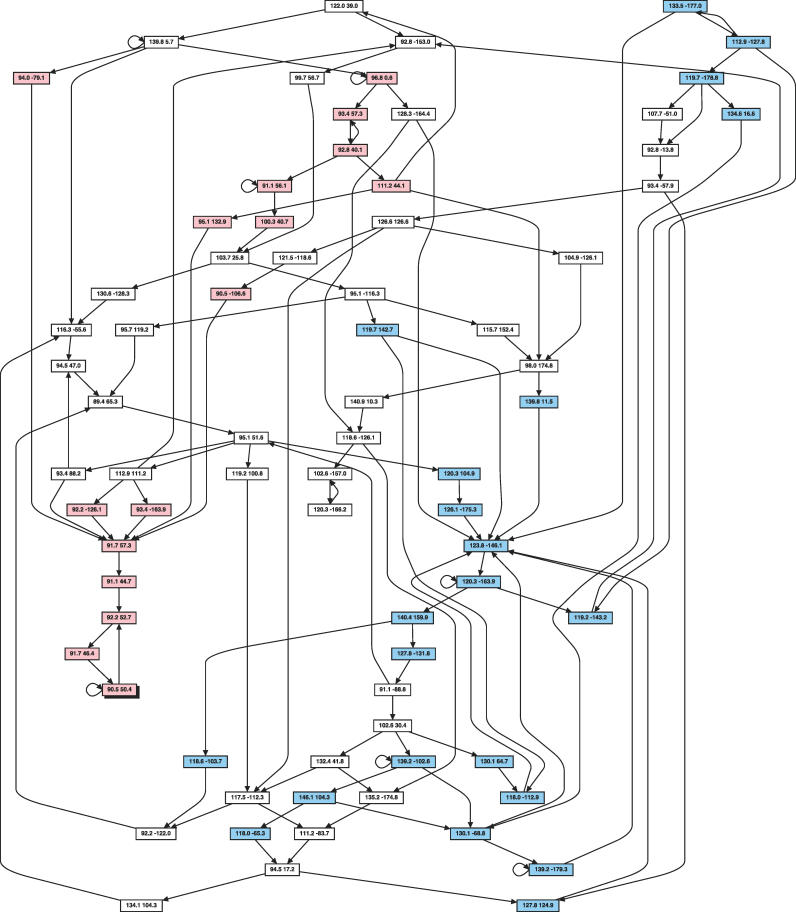
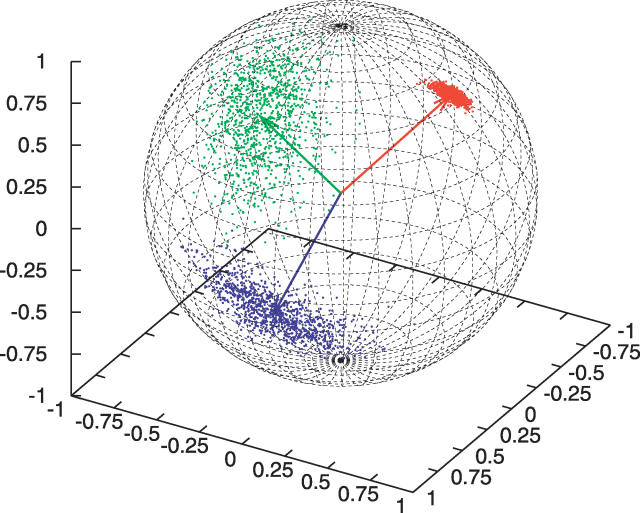

Similar articles
-
A population-based evolutionary search approach to the multiple minima problem in de novo protein structure prediction.BMC Struct Biol. 2013;13 Suppl 1(Suppl 1):S4. doi: 10.1186/1472-6807-13-S1-S4. Epub 2013 Nov 8. BMC Struct Biol. 2013. PMID: 24565020 Free PMC article.
-
Protein conformational transitions coupled to binding in molecular recognition of unstructured proteins: deciphering the effect of intermolecular interactions on computational structure prediction of the p27Kip1 protein bound to the cyclin A-cyclin-dependent kinase 2 complex.Proteins. 2005 Feb 15;58(3):706-16. doi: 10.1002/prot.20351. Proteins. 2005. PMID: 15609350
-
Toward high-resolution de novo structure prediction for small proteins.Science. 2005 Sep 16;309(5742):1868-71. doi: 10.1126/science.1113801. Science. 2005. PMID: 16166519
-
[A turning point in the knowledge of the structure-function-activity relations of elastin].J Soc Biol. 2001;195(2):181-93. J Soc Biol. 2001. PMID: 11727705 Review. French.
-
Exploring the relationships between protein sequence, structure and solubility.Curr Opin Struct Biol. 2017 Feb;42:136-146. doi: 10.1016/j.sbi.2017.01.004. Epub 2017 Feb 2. Curr Opin Struct Biol. 2017. PMID: 28160724 Review.
Cited by
-
Molecular dynamics analysis of biomolecular systems including nucleic acids.Biophys Physicobiol. 2022 Aug 23;19:e190027. doi: 10.2142/biophysico.bppb-v19.0027. eCollection 2022. Biophys Physicobiol. 2022. PMID: 36349319 Free PMC article.
-
Data-driven probabilistic definition of the low energy conformational states of protein residues.NAR Genom Bioinform. 2024 Jul 9;6(3):lqae082. doi: 10.1093/nargab/lqae082. eCollection 2024 Sep. NAR Genom Bioinform. 2024. PMID: 38984065 Free PMC article.
-
A probabilistic fragment-based protein structure prediction algorithm.PLoS One. 2012;7(7):e38799. doi: 10.1371/journal.pone.0038799. Epub 2012 Jul 19. PLoS One. 2012. PMID: 22829868 Free PMC article.
-
Calculation of accurate small angle X-ray scattering curves from coarse-grained protein models.BMC Bioinformatics. 2010 Aug 18;11:429. doi: 10.1186/1471-2105-11-429. BMC Bioinformatics. 2010. PMID: 20718956 Free PMC article.
-
Plant microRNAs: Biogenesis, Homeostasis, and Degradation.Front Plant Sci. 2019 Mar 27;10:360. doi: 10.3389/fpls.2019.00360. eCollection 2019. Front Plant Sci. 2019. PMID: 30972093 Free PMC article. Review.
References
-
- Anfinsen CB. Principles that govern the folding of protein chains. Science. 1973;181:223–230. - PubMed
-
- Levinthal C. Mössbauer spectroscopy in biological systems. Springfield (Illinois): University of Illinois Press; 1969. pp. 22–24. “How to Fold Graciously” chapter. pp.
-
- Honig B. Protein folding: From the levinthal paradox to structure prediction. J Mol Biol. 1999;293:283–293. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
